科技工作者之家
科界APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-04-01
来源:材料科学与工程
采用从头算分子动力学(AIMD)方法,研究了在现有热力学模型中假设存在缔合物的Ba-Bi液体的性质,避免了由经验势引起的误差。计算结果与文献中CALPHAD模型的计算结果吻合较好。为Ba-Bi液体中存在一个强有序的Ba4Bi3缔合物和一个弱有序的BaBi3缔合物提供了证据。
虚拟缔合被广泛地用于描述和建模具有强有序趋势的液相。然而,在大多数情况下,关于假定的缔合证据很少。近日,上海交通大学和美国宾夕法尼亚大学的Jianbo Ma等人采用从头算分子动力学(AIMD)方法,研究了在现有热力学模型中假设存在缔合物的Ba-Bi液体的性质。相关论文以题为“Anab initio molecular dynamics exploration of associates in Ba-Bi liquid with strong ordering trends”于3月21日发表在Acta Materialia上。
论文链接:
https://www.sciencedirect.com/science/article/abs/pii/S1359645420302111

最近,Lichtenstein等人通过测量钡铋合金的电动势(EMF),报道了钡(aBa)在富铋熔体中的化学活性极低。电动势研究结果表明,Ba和Bi之间存在较强的化学亲合力和短程有序,即液体中有化学缔合物。目前,有两种主要的热力学方法来解释强短程有序,即(i)采用偶对近似的准化学模型;(ii)假设液体单元素和分子类缔合物混合物的缔合模型。在缔合模型中,缔合物的组成通常与熔融温度高或混合焓为负值的金属间化合物的组成相同。缔合模型已成功地应用于二元和多组分系统的热力学模型。
Liu等人最近在对Ba-Bi体系的进行CALPHAD(计算相图)建模时,使用了液相中Ba4Bi3和BaBi3两种虚构的缔合物,这两种缔合物是根据具有两个最高同熔点的金属间化合物进行经验选择的。
基于此,研究人员利用从头算分子动力学(AIMD)模拟来研究Ba-Bi液相中缔合物的存在及其性质。AIMD方法采用基于密度泛函理论(DFT)的动态原子间相互作用计算,避免了由经验势引起的误差。结果表明,在富钡熔体中,铋原子几乎完全被钡原子所包围。在AIMD模拟中,Bi中心配位多面体与Ba5Bi3和Ba4Bi3的晶体结构密切相关,比其他多面体的寿命更长。此外,这些富Ba熔体的双Bi中心多面体通过顶点、边、面和/或双金字塔共享彼此连接,形成中程有序(MRO)。在富铋熔体中,以钡为中心的多面体也形成MROs,但它们在结构和组成上都具有多样性,寿命较短。计算结果与文献中CALPHAD模型的计算结果吻合较好。
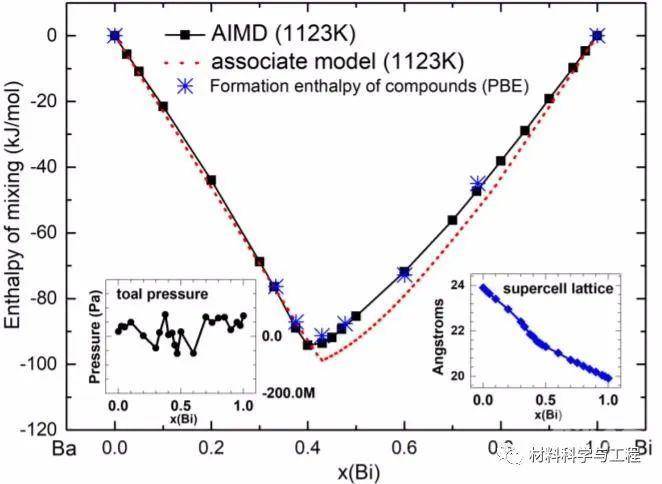 图1 利用AIMD(固体正方形,本工作)和缔合模型(点虚线,CALPHAD建模)计算的液体Ba1-xBix混合物在1123k下的焓
图1 利用AIMD(固体正方形,本工作)和缔合模型(点虚线,CALPHAD建模)计算的液体Ba1-xBix混合物在1123k下的焓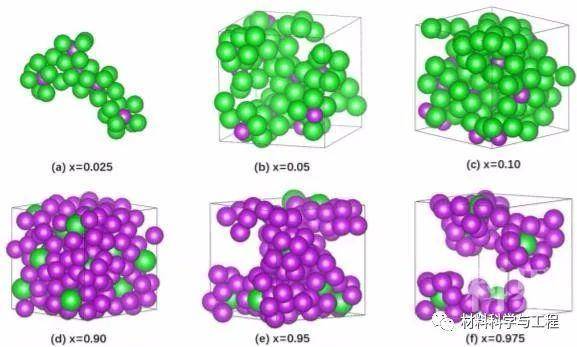 图5 在熔融Ba1-xBix中,x = 0.025 (a)、0.05 (b)、0.10 (c)、0.90 (d)、0.95 (e)和0.975 (f)的一些以Bi和Ba为中心的多面体的AIMD配置快照
图5 在熔融Ba1-xBix中,x = 0.025 (a)、0.05 (b)、0.10 (c)、0.90 (d)、0.95 (e)和0.975 (f)的一些以Bi和Ba为中心的多面体的AIMD配置快照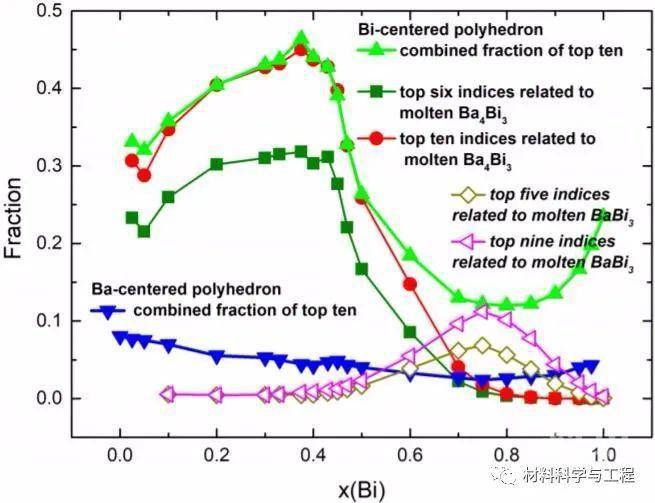 图10 主要的BIPs和BAPs的组合分数与Bi (x)摩尔分数的函数
图10 主要的BIPs和BAPs的组合分数与Bi (x)摩尔分数的函数总的来说,以上来自AIMD研究的发现为Ba-Bi液体中存在一个强有序的Ba4Bi3缔合物和一个弱有序的BaBi3缔合物提供了证据。
来源:mse_material 材料科学与工程
原文链接:https://mp.weixin.qq.com/s?__biz=MzA4NDk3ODEwNQ==&mid=2698825473&idx=5&sn=3979d7069349bad4ad7f06f1ba5e0411&chksm=baf69fd78d8116c10a7896af500f4ca9998d65eaad8bf4462db09e45f6f882e9c14078c2a73c#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
力学所提出基于双温非平衡热力学框架的非晶合金本构模型


违背“热力学原理”的日冕加热问题

基于钉扎分布原则的改进热力学磁滞模型
腐蚀过程热力学判据
地幔橄榄石中铁的氧化态及其热力学模型研究取得进展

化学热力学与绿色化学的开拓者—韩布兴
【前沿】机器翻译新突破!“普适注意力”模型:概念简单参数少,性能大增

热力学定律能指导我们减肥吗?

液体的奥秘

这么多机器学习的应用场景,金融领域到底有何不同?


科界APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号