科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-06-22
来源:宏基因组
导读临床宏基因组学(mNGS)是对患者样本中微生物和宿主遗传物质(DNA和RNA)进行综合分析的一种新兴的诊断技术,这种新兴的方法正在改变医生诊断和治疗疾病的方式,其应用范围广泛,其应用范围包括抗菌素耐药性研究、微生物组学、人类宿主基因表达(转录组学)和肿瘤学。
本文在此回顾了mNGS目前在临床和公共卫生领域的各种应用。并讨论了临床实验室实施mNGS所面临的挑战,包括验证和监管方面的考虑,并提出了克服这些挑战的步骤。最后,本文展望了临床宏基因组学领域的未来发展方向,并预测了未来5年的发展方向。
论文ID
原名:Clinical metagenomics
译名:临床宏基因组学
期刊:Nature Reviews Genetics
IF:43.704
发表时间:2019年3月27日
通信作者:Charles Y. Chiu
通信作者单位:美国加州大学旧金山分校实验医学系
综述框架
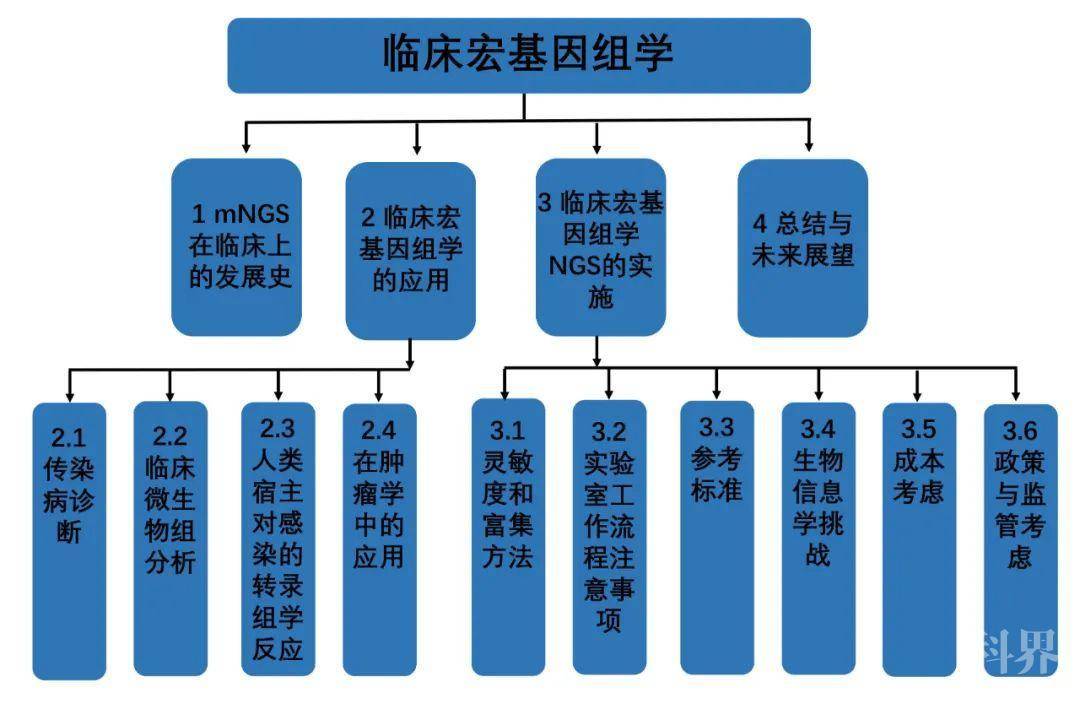
综述框架
主要内容
1 mNGS在临床上的发展史
临床微生物学领域包括诊断微生物学、从临床样本中鉴定病原体以指导感染患者的管理和治疗策略、公共卫生微生物学和监测社区内的传染病暴发源等。微生物实验室的传统诊断技术包括培养中微生物的生长和分离、病原特异性抗体(血清学)或抗原的检测以及微生物核酸(DNA或RNA)的分子鉴定,即PCR。虽然大多数分子分析只针对使用特定引物或探针的有限数量的病原体,但宏基因组方法表征了样本中存在的所有DNA或RNA,从而能够分析整个微生物组以及患者样本中的人类宿主基因组或转录体。几十年来,宏基因组方法被广泛应用于识别细菌感染,发现新的病毒病原和鉴定在健康和疾病状态下人类病毒群,以及用于法医学应用。
尽管宏基因组学具有潜力并取得了很大成功,但由于许多因素,临床诊断应用仍落后于研究进展:
1. 微生物和宿主因素的复杂相互作用影响着人类的健康,例如微生物在调节宿主免疫反应中的作用,通常不清楚被检测到的微生物是污染物、定殖者还是真正的病原体。
2. 此外,通用参考标准并且缺乏证明临床宏基因组分析的试验验证、重复性和质量保证的有效方法。
3. 考虑到成本、报销、周转时间、监管因素,也许最重要的是,临床效用也是在患者护理环境中常规实施临床mNGS的主要障碍。
本文在此回顾了mNGS目前在临床和公共卫生领域的各种应用。并讨论了临床实验室实施mNGS所面临的挑战,包括验证和监管方面的考虑,并提出了克服这些挑战的步骤。最后,展望了临床宏基因组学领域的未来发展方向,并预测了未来5年的发展方向。
2 临床宏基因组学的应用
到目前为止,临床宏基因组学的应用包括各种综合征和样本类型的传染病诊断,疾病和健康状态下的微生物组分析,以及人类宿主对感染的转录组学反应及肿瘤相关病毒及其基因组整合位点的鉴定(图1)。除传染病诊断外,临床实验室采用mNGS的速度很慢,大多数应用还没有纳入常规临床实践。尽管如此,这些应用的广度和潜在临床实用性很可能在不久的将来改变诊断微生物学领域。
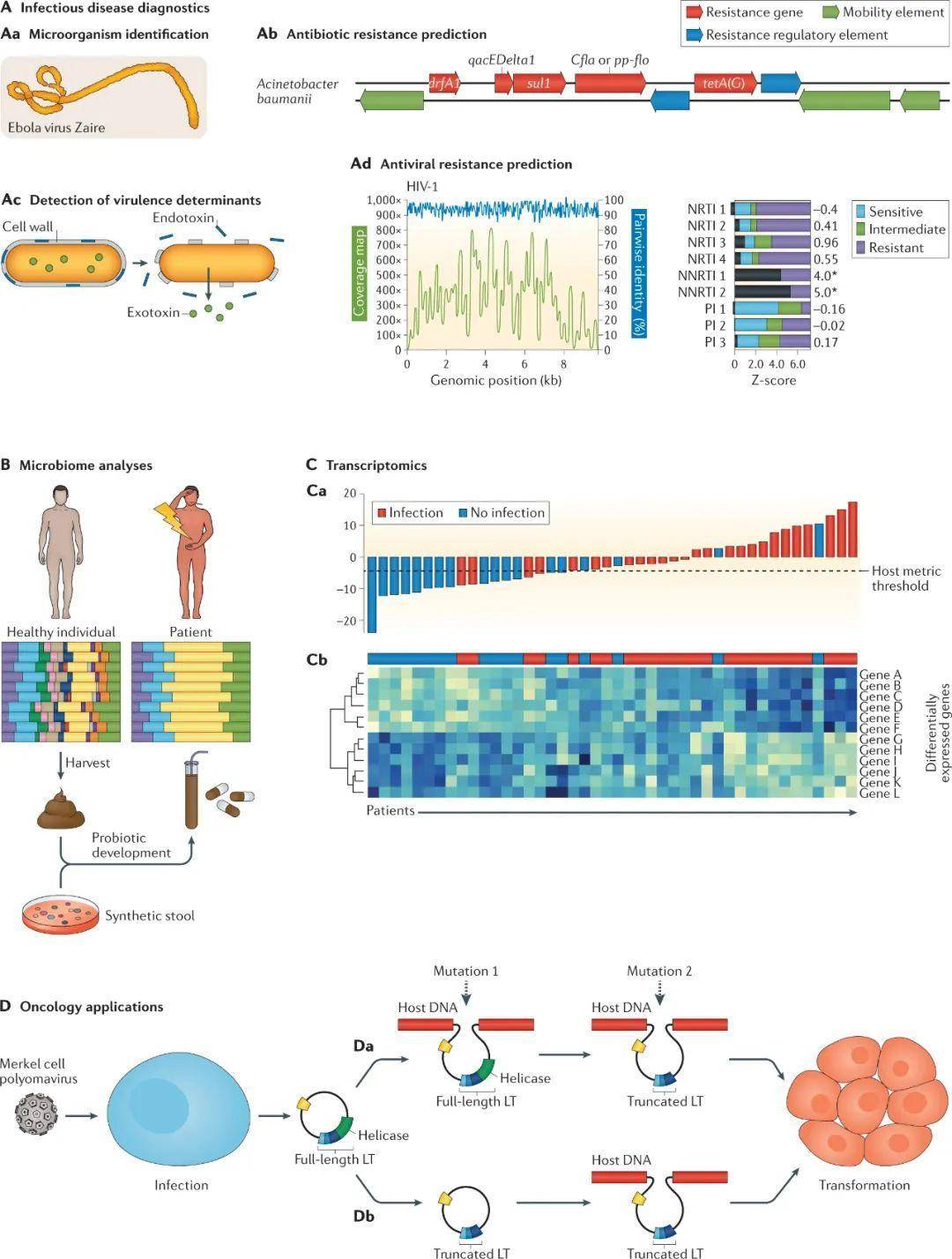
图1 宏基因组测序的临床应用。(A)在传染病诊断中的应用包括直接鉴定原始临床样本中的微生物(Aa);通过鉴定耐药基因预测抗生素耐药性(Ab);检测物种或菌株水平的毒力决定因素,如分泌特定的内毒素或外毒素(Ac);抗病毒抗性预测(Ad)。(B )微生物组分析可在急性和慢性疾病状态下告知疾病预后,并为益生菌疗法的发展奠定基础。(C) RNA测序的转录组学可以在人类宿主反应的基础上提高对感染性和非感染性疾病的诊断水平。利用NGS进行宿主转录组分析,可以构建一个分类指标,以高精度区分感染患者和未感染患者(Ca)。聚类热图分析确定了与感染相关的个体差异表达的宿主基因和与未感染相关的宿主基因(Cb)。(D)在肿瘤学中,病毒性肿瘤测序或液体活检分析可用于同时检测病原体和鉴定宿主基因突变。mNGS可用于检测Merkel细胞多瘤病毒,该病毒与Merkel细胞癌的发生有关。宿主DNA的同时测序可以识别由包含T抗原(LT)的病毒基因组整合、随后的LT抗原(Da)或病毒基因组整合前的LT抗原(Db)引起的突变。这两种突变导致细胞转化,从而推动肿瘤增殖。虽然这些基于测序的应用很有前途,但其中许多已经被纳入常规临床实践中。
2.1 传染病诊断
传统的诊断病人感染性疾病的临床模式是由医生制定鉴别诊断,然后进行一系列测试,试图找出病因。临床样品中病原体常规检测的范围包括鉴定培养中生长的微生物(例如,通过生化表型检测或基质辅助激光解吸/电离(MALDI)飞行时间质谱法),检测生物体特异性生物标记物(例如抗原用乳胶凝集试验或酶联免疫吸附试验(ELISA)抗体试验或用PCR对单个试剂进行核酸试验。这些通常包括与确定的临床综合征有关的最常见病原体,如脑膜炎和脑炎、急性呼吸道感染、败血症或腹泻病。
分子诊断分析为诊断最常见的感染提供了一种相当经济有效和快速的方法(通常< 2 h的周转时间)。然而,目前使用的几乎所有常规微生物试验一次只能检测一种或有限的病原体,或者要求从临床样本中成功培养微生物。相比之下,虽然目前使用的NGS分析方法在速度上无法与传统的检测方法相比——在标准的Illumina仪器上单独进行的测序需要超过18小时——但mNGS可以使多种病原体——病毒、细菌,真菌和/或寄生虫——根据唯一可识别的DNA和/或RNA序列从培养物或直接从临床样本中识别。NGS方法的另一个关键优点是,测序数据可能被用于除了鉴定致病病原体之外的其他分析,例如通过RNA测序(RNA-seq)对转录组进行分析,从而确定微生物组特征和人类宿主反应的平行分析。
因此,NGS在诊断中的临床应用可能是在最难诊断的病例中,或者对于潜在病原体谱较大的免疫功能低下的病人。最终,mNGS可能与多重分析具有成本竞争性,或用作排除感染性病因的预先“排除”分析。当然,通过多重PCR检测或NGS检测核酸,本身并不能证明被鉴定的微生物是致病的原因,而且这些发现必须在临床上加以解释。特别是,在临床样本中发现非典型或新的传染源随后进行验证性研究,如组织活检样品的正交试验和血清转化证明,或酌情使用细胞培养或动物模型,以确定其真正的致病潜力。在研究实验室或临床实验室进行的临床样品的NGS涉及许多步骤,包括核酸提取、DNA和/或RNA富集、文库制备、PCR扩增(如果需要)、测序和生物信息学分析(图2)。任何能产生足够核酸的体液或组织都可以进行NGS分析,这种分析既可以是靶向的,也可以是丰富单个基因或基因组区域的,即非靶向的,就像宏基因组“鸟枪”方法一样(图2)。具体步骤的细节因实验室而异,在其他地方有详细描述。
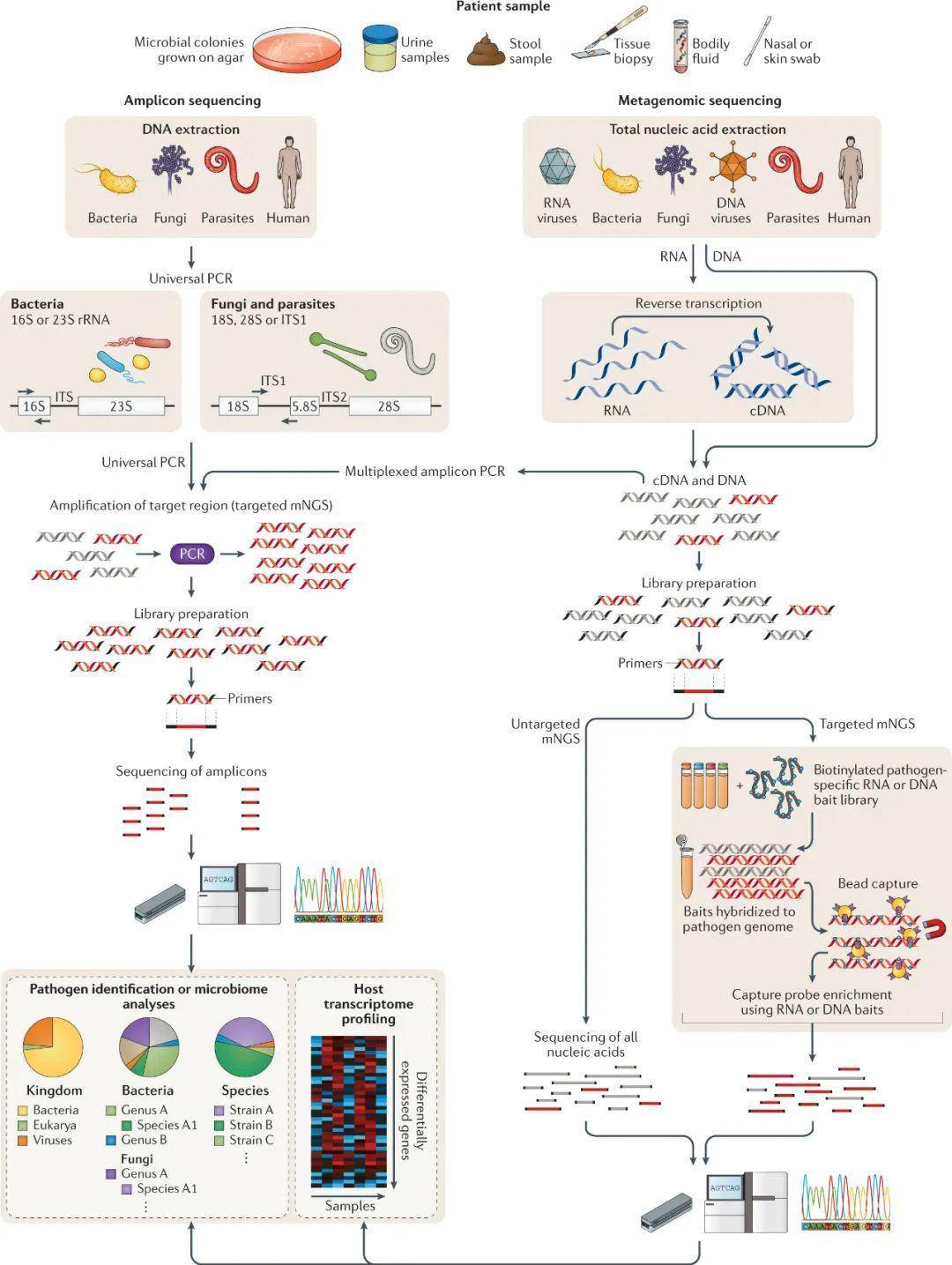
有针对性的方法有利于增加序列数据中病原体读取的数量和比例。这一步骤可以提高对目标微生物的检测灵敏度,尽管它限制了可以识别的潜在病原体的广度。
1. 靶向方法的一个例子是使用高度保守的引物对临床样本中与特定类型相对应的所有微生物进行通用PCR扩增和检测,例如对细菌18S rRNA进行16S核糖体RNA(rRNA)基因扩增(图2)。靶向NGS方法的另一个例子是设计横跨基因组的引物,以促进PCR扩增和扩增,从而直接从临床样本中恢复病毒基因组。该方法已被用于跟踪美国寨卡病毒(ZIKV)和西非埃博拉病毒的演变和传播,并展示了实时监测对公共卫生干预措施的影响。
2. 另一种有针对性的方法是捕获探针富集,即利用捕获“诱饵”探针对宏基因组文库进行杂交。这些探针的长度一般为30-120 bp,探针的数量可以从小于50到大于200万不等。尽管这种富集方法已被证明在研究环境中提高了宏基因组检测的灵敏度,特别是对于病毒,但它还没有被常规用于临床诊断。这一方法的一个有希望的应用可能是丰富临床样本以表征抗生素耐药性,这是医院中的一个相当大的问题,也是美国国家打击抗生素耐药性细菌行动计划的主要重点。然而,与非靶向传染病诊断方法相比,捕获探针富集的缺点包括偏向于靶向微生物、增加步骤、增加成本和由于最大效率所需的额外处理而导致杂交时间长(24-48小时)。
2.1.2 非靶向宏因组NGS分析
非靶向mNGS分析放弃使用特异性引物或探针。相反,整个DNA和/或RNA(在反向转录到cDNA之后)被测序。利用细菌或真菌的纯培养物,mNGS-reads可以组装成部分或完整的基因组。这些基因组序列随后被用于亚型和/或监测医院爆发,以支持感染控制和/或公共卫生监测工作。
临床样本的非靶向mNGS可能是综合诊断感染最有希望的方法。原则上,几乎所有的病原体,包括病毒、细菌、真菌和寄生虫,都可以在一次检测中识别。mNGS的一个局限性是,该方法的灵敏度严重依赖于背景水平。例如,与无细胞体液相比,组织增加了人类宿主背景,导致微生物读长的数量和比例减少,从而降低了对mNGS的敏感性。此外,定义诊断或预测疾病发展的特定微生物图谱可能很困难,特别是在含有复杂微生物群的非灭菌部位,如呼吸道分泌物或粪便。尽管如此,有几个小组已经在临床实验室改进修正案(CLIA)中成功地验证了mNGS——经认证的诊断感染的临床实验室,包括脑膜炎或脑炎、脓毒症和肺炎,这些分析现在可用于患者的临床参考试验。
2.2 临床微生物组分析许多研究人员现在使用mNGS而不是16S rRNA基因的靶向测序来深入鉴定微生物组。公众越来越认识到微生物组及其可能参与急性和慢性疾病的状态。然而,目前还没有一种基于微生物组学的检测方法被临床用于疾病的诊断或治疗,部分原因是人们对微生物组学的复杂性及其在疾病发病机制中的作用认识不足。
微生物组学分析在Clostridium difficile 相关疾病的治疗和治疗中有着广阔的应用前景。C. difficile 能感染肠道,产生毒素,可导致腹泻、脱水、败血症和死亡。C. difficile 难治性感染只发生在微生物群的环境中,而微生物群的环境会因暴露于广谱抗生素或最近的胃肠道手术等因素而改变。粪便移植治疗和潜在治疗C. difficile 感染的有效率为80%-90%,这突出了微生物群在艰难梭菌感染中的重要性。在多个研究中使用mNGS来表征微生物组分有助于细菌-益生菌混合物的开发,这种混合物可以作为预防或治疗艰难梭菌相关疾病的药丸使用(图1b)。
微生物组的另一个潜在应用是分析细菌多样性,这可以提供病人的疾病是传染性的还是非传染性的线索。例如,一项用于鉴别肺炎患者呼吸道病原体的mNGS研究发现,经培养证实感染的个体其呼吸道微生物组的多样性显著降低。这种被称为失调的微生物群的改变也被证明与对于肥胖、糖尿病和炎症性肠病,微生物群的操作可能是治疗这些病理状况的一个途径。
2.3 人类宿主对感染的转录组学反应
临床mNGS通常侧重于微生物丰度;然而,在研究人类宿主对感染的反应中,基因表达分析具有互补作用(图1C)。用于检测病原体(如临床样本中的RNA病毒)的RNA文库的mNGS偶然产生用于转录组(RNA-seq)分析的宿主基因表达数据。尽管RNA-seq分析通常是在全血或外周血单个核细胞(PBMC)样本上进行的,但任何体液或组织类型都可能适合这些分析。通过RNA-seq表达谱对基因进行分类已被用于描述几种感染,包括葡萄球菌菌血症、莱姆病、念珠菌病、结核病(区分潜在和活动疾病风险)和流感。基于机器学习的RNA-seq数据分析已被用于癌症分类,这些方法的翻译可能有希望用于传染病。含有数量有限的宿主生物标记物正在发展成为流感、结核和细菌性败血症的诊断分析。
虽然迄今为止还没有一种基于RNA-seq的分析方法被临床证实可用于患者,但RNA-seq分析的潜在临床影响是很高的。从与活性微生物基因表达相对应的微生物中提取的RNA的询问可能能够区分感染与定殖和活组织与死组织。此外,人类宿主的RNA-seq分析可用于直接从临床样本中识别新的或未被充分认识的宿主-微生物相互作用,如之前对莱姆病、登革热或疟疾患者所示。RNA-seq在病原菌仅短暂存在的临床病例中可能特别有用(例如早期莱姆病或虫媒病毒感染,包括西尼罗河病毒);类似于血清学检测,根据病原体特异性人类宿主反应,间接诊断感染是可能的。对病原体特异性宿主反应的分析也可能有助于在复杂的临床亚基因组样本(如多微生物脓肿或呼吸道流感)中鉴别真正的病原体。RNA-seq的另一个有前途的应用是在区分急性疾病的传染性和非传染性病因。例如,如果根据宿主反应判断一种疾病更可能是非传染性的(例如,自身免疫性疾病),临床医生可能更愿意停止使用抗生素,并用类固醇和其他免疫抑制药物积极治疗患者。随着大规模测序数据的不断产生,也许是由常规的临床mNGS测试驱动的,对人类阅读的二次挖掘可能通过合并微生物和宿主基因表达数据来提高临床诊断的准确性。
2.4 在肿瘤学中的应用
在肿瘤学中,全基因组或定向NGS方法识别突变基因可用于同时发现与癌症相关的病毒(即疱疹病毒、乳头状瘤病毒和多瘤病毒)和/或收集病毒相互作用的数据。例如,mNGS对发现Merkel细胞多瘤病毒(图1D)至关重要,目前认为Merkel细胞癌是一种罕见的皮肤癌,最常见于老年患者。迄今为止,美国食品和药物管理局(FDA)已批准临床使用两个NGS小组测试肿瘤样本中可操作的基因组畸变。在这些样本中检测与整合病毒和外源病毒相对应的读码,可以在面板上添加特定的病毒探针,也可以在对整个肿瘤基因组或菲氏体进行测序时偶然完成。
对癌症中的综合性或活动性病毒感染及其参与信号传导途径的额外了解,可为靶向抗病毒和/或化疗药物的预防和治疗干预提供信息,这一点可通过治疗后丙型肝炎病毒相关性肝细胞癌的风险降低来证明直接作用的抗病毒药物。在未来,液体活检标本(如血浆)中无细胞DNA的mNGS可用于免疫功能低下患者早期癌症的同时鉴别和感染的诊断
3 临床宏基因组学NGS的实施
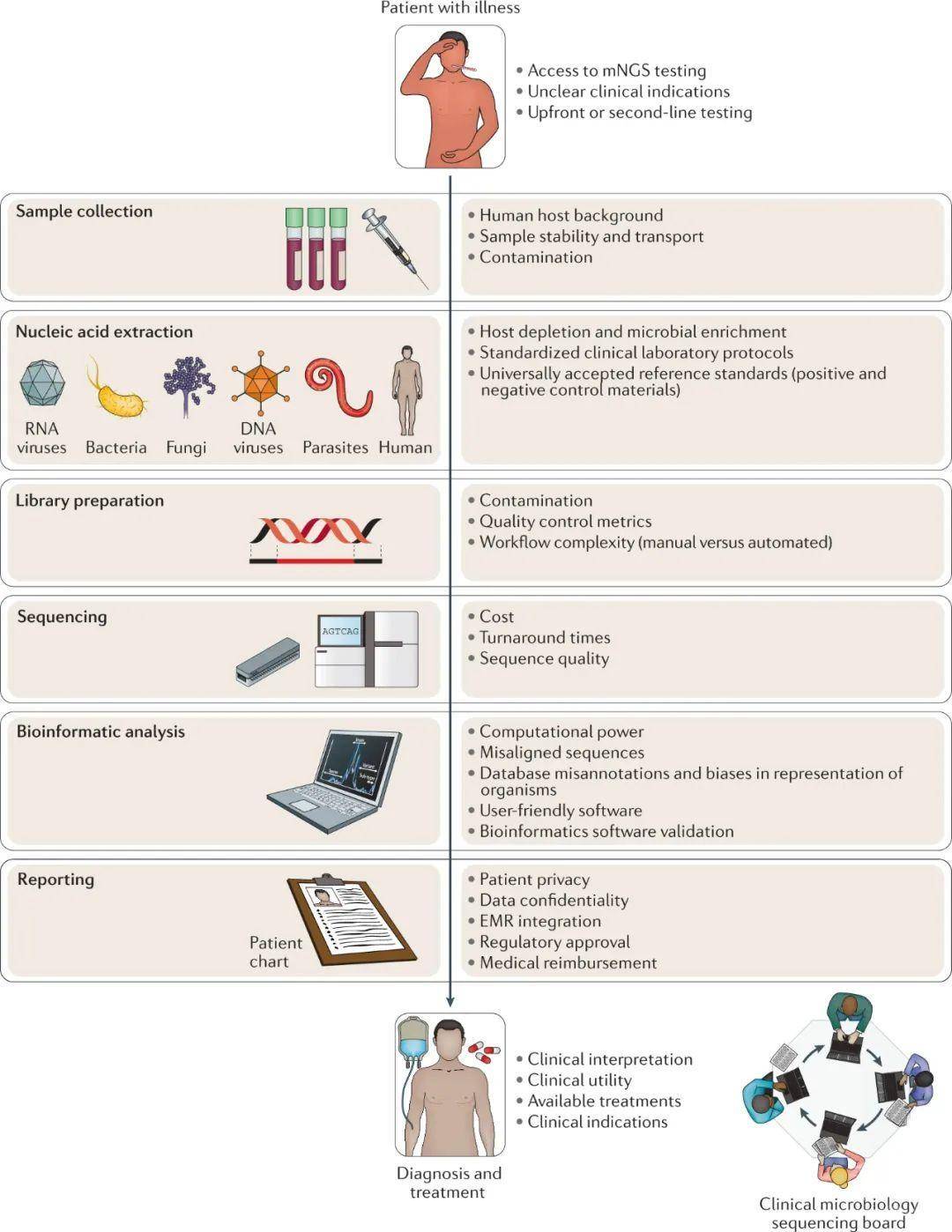
在临床实验室实施mNGS是一项复杂的工作,需要使用符合监管标准的质量管理方法定制研究方案。准备试剂、测序仪器和生物信息学工具在研究环境中不断变化。然而,在临床实验室中,分析需要遵循标准化协议来实施。对的任何组件所做的更改在对患者进行检测之前,需要对检测进行验证并证明其具有可接受的性能。定期更新和重复验证研究被认为是必要的,以纳入NGS试剂、协议和仪器的临时技术进步。
病原体检测的宏基因组方法对于临床验证来说是一个特别具有挑战性的场景(图3),因为对于要被认为是有效的分析来说,对本质上无限数量的不同生物体进行测试是不现实的。尽管FDA已经为NGS感染性疾病试验的临床验证提供了一般指南,但是对于mNGS试验的临床实施没有明确的建议,也没有提到具体的要求。然而,可以采取一种最佳实践方法,包括使用具有代表性的有机体进行失效模式分析和性能特性评估,并持续进行分析监测和独立确认意外结果。
3.1 灵敏度和富集方法
mNGS的一个关键限制是其在高背景下的敏感性降低,主要来自人类宿主(例如,在组织活检中)或微生物组(例如,在粪便中)。该背景可能与临床相关,因为感染的病原体负荷可能非常低。
已开发出用于RNA文库的宿主缺失方法,并证明其有效性,包括提取后的DNase I处理以去除残留的人类背景DNA;使用RNA探针然后进行RNase H处理;抗人类和线粒体rRNA抗体(临床样本中最丰富的宿主RNA类型);和/或基于CRISPR-Cas9的方法。
遗憾的是,目前还没有比较有效的DNA文库并行方法。使用抗甲基化人类宿主DNA的抗体可以在3-5倍的范围内实现有限的富集,因为在大多数病原体基因组中缺乏甲基化的DNA,从而丰富了微生物的序列。人类细胞的差异裂解,然后用DNase Ⅰ降解背景DNA,从而保留和富集有细胞壁的生物体(包括一些细菌和真菌)中的核酸,已被证明可提供高达1000倍的微生物富集量。然而,微分分析方法的性能会受到许多因素的限制。这些局限性包括:对没有细胞壁的微生物,如支原体或寄生虫的敏感性可能降低;使用额外的试剂可能会使外源性背景污染增加,这可能是矛盾的;以及无法从人体宿主在体内溶解的死生物体中检测游离核酸免疫细胞或抗生素治疗。
在某种程度上,人类宿主背景的限制通过增加可用测序器的容量来实现。例如,通过对脑组织进行超深测序,在一名脑炎患儿身上检测到一种星形病毒,在约1.34亿次检测中,仅检测到1612次(0.0012%)序列。另一种提高灵敏度的方法是利用一种混合方法进行富集,例如用带尖峰引物的宏基因组测序。结合靶向测序和非靶向测序,该方法使用可变大小的短引物板(100-10000)添加到反应混合物中以富集特定靶生物,同时保留宏基因组测序广度。当反向转录时,一组ZIKV特异性引物被发现可使ZIKV的读写次数增加10倍以上,而不会明显降低对其他病原体的广泛的亚基因组敏感性,使全基因组病毒测序能够确定ZIKV从巴西传播到中美洲和墨西哥的特征。
3.2 实验室工作流程注意事项
mNGS分析的复杂性要求在样品处理方面有训练有素的人员和极为谨慎的态度,以避免错误和交叉污染。即使是在样本采集、等分、核酸提取、文库制备或汇集过程中引入的微量外源DNA或RNA,也会从污染的读长中产生可检测的信号。此外,实验室表面、消耗品和试剂都不是不含DNA的。通常需要维护在mNGS数据中检测到的、由正常菌群或实验室污染引起的背景微生物数据库,以便进行准确的mNGS分析。
临床实验室操作的特点是有一个确定的工作流程和预定的人员配置水平,并且比研究实验室更不适合按需测试。由于样品通常是分批处理的,分批分析的频率是总周转时间的主要决定因素。除非完全自动化的样品处理系统是现成的,实验室操作mNGS需要相当多的动手时间来执行,以及临床工作人员谁是在分子生物学程序的高度培训。对于重复性的工作,如移液管,存在人体工程学的问题,也可能会无意中混淆样本或遗漏工作流程中的关键步骤。在复杂的mNGS中保持高质量程序可能会给工作人员带来压力,因为样本处理中的轻微偏差可能导致产生的结果发生重大变化。通过轮班将分析工作流程分为多个离散步骤,有助于避免实验室误差。
3.3 参考标准
需要特征良好的参考标准和控制,以确保随着时间的推移mNGS分析质量和稳定性。大多数可用的宏基因组参考材料高度定制以用于特定应用(例如,酶生物学微生物群落标准用于微生物组分析和细菌和真菌亚基因组)和/或聚焦于更有限的生物体谱(例如,国家标准与技术研究所(NIST)混合微生物DNA检测参考材料,其中仅包含细菌)。因此,这些材料可能不适用于非靶向的mNGS分析。
可以开发由一个微生物库(模拟微生物群落)或其核酸组成的定制混合物作为外部控制,以确定mNGS检测的检测极限。内部的顶级控制标准可用于其他NGS应用,如RNA-seq的转录组分析,External RNA Controls Consortium(ERCC)RNA标准由跨越一系列核苷酸长度和浓度的合成RNA寡核苷酸组成。整套或部分ERCC RNA标准(或其DNA等效物)可用作内对照,以控制分析抑制,并通过标准曲线分析量化检测到的病原体的滴定度。然而,由于缺乏公认的mNGS参考标准,很难比较不同实验室之间的分析性能。迫切需要标准化的参考生物体和基因组材料来促进这种比较,并确定最佳的分析方法。
3.4 生物信息学挑战
用于分析mNGS数据的用户友好的生物信息学软件目前仍然没有。因此,用于临床mNGS数据分析的定制生物信息学流程仍然需要训练有素的编程人员来开发、验证和维护用于临床的需要。实验室可以在本地托管计算服务器,也可以将生物信息学分析和数据存储转移到云平台。在这两种情况下,硬件和软件设置都可能很复杂,必须采取适当措施保护患者序列的机密数据和信息,特别是在云环境中。测序数据的存储需求会很快变得相当大,临床实验室必须决定数据存储的数量、位置和持续时间。
用于mNGS分析的生物信息学流程使用许多不同的算法,通常是为研究环境开发的,并且由软件开发人员不断更新。对于实验室程序,通常需要对软件进行自定义修改,然后锁定软件和参考数据库以进行临床验证。典型的生物信息学由一系列来自原始输入FASTQ文件的分析步骤组成,包括质量和低复杂度过滤、接头序列剪切、人类宿主移除、通过与参考数据库对齐进行微生物识别、可选的序列组装和分箱的分类,在诸如科、属和种的水平上的连续序列(图4)。必须仔细评估流程中的每个步骤,以确保数据处理的准确性和完整性,同时考虑误差的传播。敏感性分析应包括电子数据和临床样本数据。可以准备定制的数据集,以模拟输入序列数据,并扩大检测到的微生物的范围。使用标准化参考资料和NGS数据集也有助于不同生物信息学流程的比较评估。
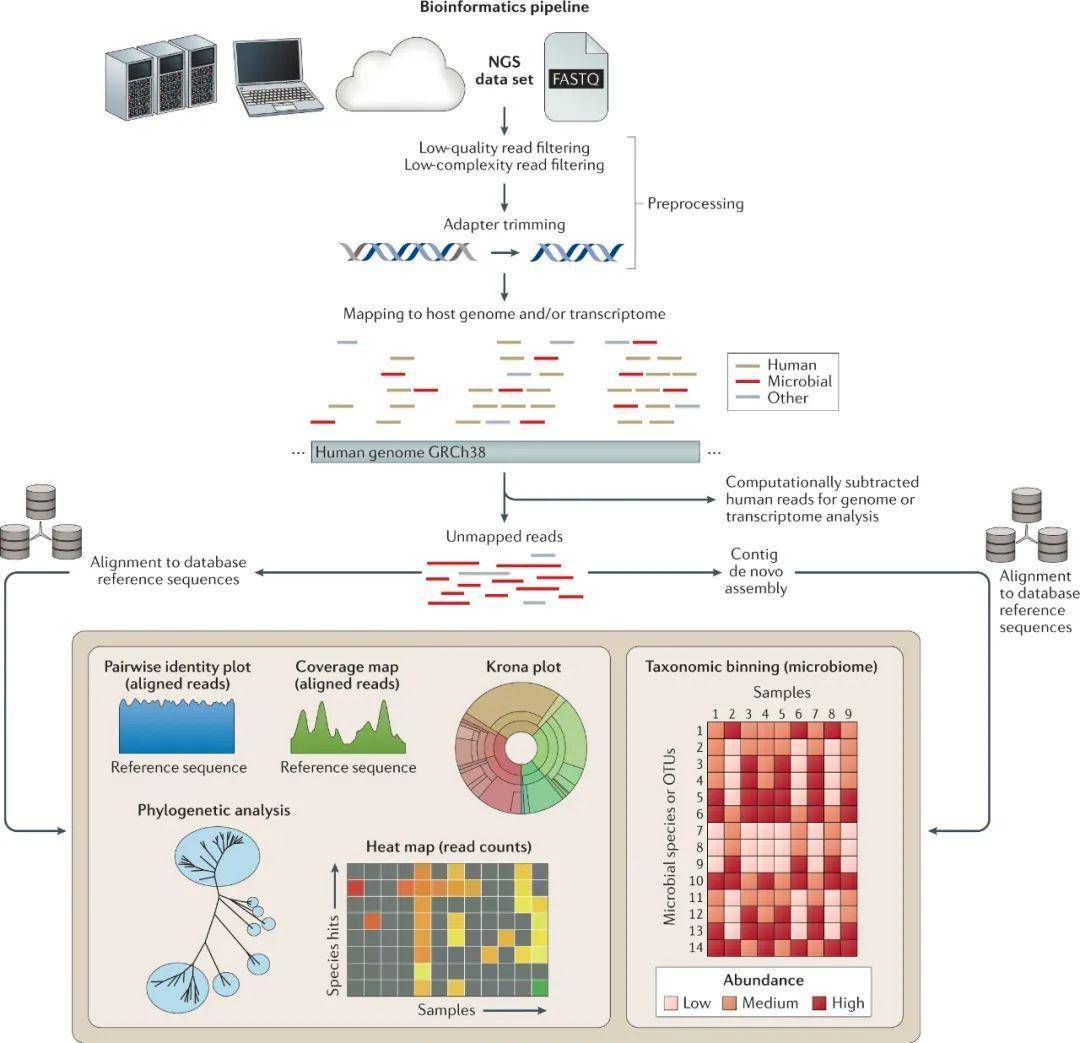
此外,微生物参考基因组的公共数据库正在不断更新,实验室除了处理潜在的错误注释和其他数据库错误外,还需要跟踪所使用的确切版本。包含公开存放序列的更大、更完整的数据库,如National Center for Biotechnology Information(NCBI)核苷酸数据库,比FDA-Argos或FDA Reference Viral Database (RVDB)等更精确、更有限的数据库更全面,但也包含更多错误。一种结合多个数据库注释序列的组合方法可以提高微生物鉴定的敏感性和特异性。
生物信息学分析的性能验证是一项耗时的工作,包括对照组和患者数据集的分析和比较,以及用正交临床试验确定最终结果的准确性。建立阈值可以将真阳性匹配从背景中分离出来,并且这些阈值可以包含诸如与检测到的微生物对比序列读长数、标准化为百万读长数、外部无模板控制样本或内部标准材料等度量;覆盖的非重叠基因组区域的数量;以及相对于阴性对照样品的临床样品的读长丰度(以避免报告污染生物体)。受试者曲线(ROC)分析是一种有用的工具,可以确定具有已知结果的临床样本训练集的最佳阈值,并使用独立的验证集验证预先设定的阈值
在实验室工作流程中,分析软件和参考数据库最好在验证和临床使用前确定。许多实验室同时维护临床参考数据库的生产版本和最新开发版本(例如,NCBI核苷酸数据库每两周更新一次),数据库定期按预先指定的间隔更新。应使用标准化数据集在任何更新后验证数据库,并确保分析结果准确且可重复,因为新保存的序列和临床元数据可能会引入错误。
3.5 成本考虑
虽然在序列数据的生成方面已经有了实质性的成本降低,但是用于测序的每个样本试剂的总成本仍然相当高。大多数实验室缺乏自动设备在一次运行中多路传输大量患者样本的自动化协议。因此,大多数mNGS的库准备方法都是手动执行的,因此需要大量的工作人员时间。运行和维护生物信息学分析流程的成本也相当可观,为确保监管监督而采取的措施也会显著增加成本。这导致每个分析样本的总成本为数百到数千美元,比许多其他临床试验的成本都要高。
mNGS样品处理需要在硬件上进行技术改进,以提高吞吐量和降低成本。随着NGS程序变得更加标准化,随着液体处理生物机器人的使用,自动化程度不断提高。通常情况下,临床MNG需要两个生物设备来完成前扩增和后扩增步骤,以避免PCR扩增子交叉污染。随着最新一代测序仪(如Illumina NovaSeq instruments)的输出大大增强,增加混样测序也是可能的。但是,由于批量处理的要求以及样本工作流程和计算分析的考虑,每次运行大量样本的潜在限制是临床使用的总周转时间更长。此外,只有在参考实验室中才能对NGS的临床样品进行高通量处理。用于NGS样品库制备的微流控装置,如VolTRAX,最终可以使临床医生在医院实验室或护理点更广泛地使用mNGS。
3.6 政策与监管考虑
临床实验室受到高度监管,一般实验室和测试要求适用于为患者护理报告的所有分子诊断分析。质量控制是最重要的,必须开发方法以确保整个分析工作流程的分析精度。重要的质量控制步骤包括初始样本质量检查、库参数(浓度和大小分布)、序列数据生成(聚类密度和Q-分数)、内部控制的恢复和外部控制的性能。应记录分析开发和实施过程中产生的验证数据,并提供给实验室检查员(用于实验室开发的测试)或提交给监管机构,如美国的FDA或欧洲的欧洲药品管理局(EMA),以供批准。
持续的监测对于mNGS分析尤其重要,它可以随着时间的推移验证可接受的性能,并调查非典型的发现。监测是通过样本内部控制、运行中控制样本、污染刷卡测试和定期能力测试来完成的。通过回顾患者的临床图表或使用正交法进行实验室验证试验,进一步调查意外或异常结果。实验室对以前未经鉴定的微生物的鉴定,一般应通过临床参考或公共卫生实验室检测进行独立鉴定。应评估非典型或新生物的临床意义,并应报告这些发现,并与卫生保健提供者讨论,同时考虑其潜在的致病性以及进一步的检测和治疗选择。临床微生物测序委员会,以肿瘤学中的肿瘤委员会为模型,可以通过实时电话会议召集,在临床环境中与治疗提供者讨论mNGS结果(图3)。如发现有公共卫生影响的微生物,如Sin Nombre汉坦病毒或埃博拉病毒,应酌情向相关公共卫生机构报告。
4 总结与未来展望
文库的制备方法、序列生成和计算生物信息学方面的技术进步,使以较低成本进行更快速和更全面的宏基因组分析成为可能。测序技术及其应用不断发展。实时测序技术在临床医学和公共卫生中的应用可能是一种突破的技术,因为实验室已经开始应用这些工具来诊断非典型感染和跟踪病原体的爆发。
尽管如此,在实施常规病人护理的mNGS时,仍然存在巨大的挑战。特别是,在高核酸背景或极低病原体滴度的临床样本中,病原体检测的灵敏度降低;这一问题是随着成本持续下降,增加每个样本的测序深度只能部分缓解这种情况。作为一种综合性的直接检测方法,mNGS可能最终取代临床微生物学中的培养、抗原检测和PCR方法,但病毒血清学检测等间接方法将继续在感染诊断工作中发挥关键作用,而功能分析,如培养和表型敏感性测试,将可能永远有用的研究研究。总之,虽然目前的局限性表明,mNGS不太可能在短期内取代传统的诊断方法,但在某些临床情况下,它可能是一种补充性的,也许是必要的测试。
尽管在多个病例报告和小病例系列中已证明使用mNGS通知临床护理,但几乎所有的研究都是回顾性的,临床实用性尚未在大规模前瞻性临床试验中得到证实。前瞻性临床研究对于了解何时进行mNGS以及如何将诊断结果与其他方法进行比较至关重要。例如,mNGS转录组学方法可能实现有效的治疗分类,即仅对显示基因表达“传染性特征”的患者需要抗菌药物,而具有“非传染性特征”的患者可因其他原因进行治疗。特别是,需要前瞻性临床试验和经济数据来证明这些相对昂贵的试验在改善患者预后方面的成本效益,以证明其使用的合理性。这些数据还将为监管机构批准和临床报销提供途径。高质量的证据表明,临床亚基因组分析在指导患者管理方面是有效的,这需要尽可能减少潜在分析和患者选择偏差,并使用大患者队列生成的数据集比较相关的健康结果。
来源:meta-genome 宏基因组
原文链接:https://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247490649&idx=2&sn=cc5556acd3d2196aa851740c8a72509a&chksm=fab9f6e8cdce7ffed6538a7fbf880ce3de7cdc6c6a3fda039484aa9605f232258a95a5b51590#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
辽宁省北票市结核病防治所举办2016年学校结核病防治知识培训班


生物学: 咖啡害虫的致命弱点
爱心汇聚力量,团结战胜灾害
机体感染病原体之前 肠道微生物或会塑造机体多种抗体的产生!
疾病: 了解风湿性关节炎的发病机制

【院士之家】中国工程院院士徐建国: 未来新发传染病的挑战是应对新的病原体

Nature返本还原!人胎盘没有微生物组,但可能有潜在病原体

Nature:基于宏基因组测序构建人类肠道微生物组参考基因集

基于宏基因组测序技术解析坛紫菜附生微生物菌群 | JOL论文推荐
演化:病原体有时对女性更宽容


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号