科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-08-04
来源:宏基因组
功能预测在基于16S rRNA扩增子的研究中,因为了解微生物群落功能的需要,我们经常会使用一些软件工具对16S rRNA扩增子数据进行功能预测,比如PICRUSt2。
这些功能预测的结果大多是基于KEGG数据库,KEGG是一个整合了基因组、化学和系统功能信息的数据库。
把从已经完整测序的基因组中得到的基因与更高级别的细胞、物种和生态系统水平的系统功能关联起来是KEGG数据库的特色之一。
与其他数据库相比,KEGG 的一个显著特点就是具有强大的图形功能,它利用图形而不是繁缛的文字来介绍众多的代谢途径以及各途径之间的关系,这样可以使研究者能够对其关注的代谢途径有更直观全面的了解。
KEGG GENES数据库提供关于在基因组计划中发现的基因和蛋白质的序列信息。
KEGG PATHWAY数据库包括各种代谢通路、合成通路、膜转运、信号传递、细胞周期以及疾病相关通路等,此外还收集了各种化学分子、酶及以及酶促反应等相关信息。
KEGG Module数据库是 KEGG 收集的一系列的功能单元,用于基因组注释和生物学解释。
KEGG Orthology (KO)系统通过把分子网络的相关信息连接到基因组中,提供了跨物种注释流程。
ko:表示通路,这个通路是不分物种的,相当于所有物种某一功能步骤的并集。
KEGG 数据库包含 8 类生物代谢通路的分析:
Global Map
Metabolism, Genetic Information Processing
Environmental Information Processing
Cellular Processes
Organismal Systems
Human Diseases
Drug Development
在每一个大类的代谢通路中,被系统分类为二、三、四层。
第二层目前包括有 39 种子功能;第三层即为其代谢通路图;第四层为每个代谢通路图的具体注释信息。
丰度分布图绘制使用PICRUSt2等功能预测工具得到的KEGG路径预测结果同样是有层级的,当我们想要知道样本中微生物群落的功能主要涉及哪些路径时,就需要对这些具有层级的功能预测结果进行绘图。
一般情况下在图像中会展示第一和第二层级,因为三、四层级条目过多,在一幅图中进行展示也看不清,通常会挑选感兴趣的特定功能单独呈现。
绘图数据绘图数据是一个简单的三列表格,包括第二层级的39个代谢路径及其对应的第一层级信息,还有就是每个代谢路径在所有样本中的平均相对丰度。
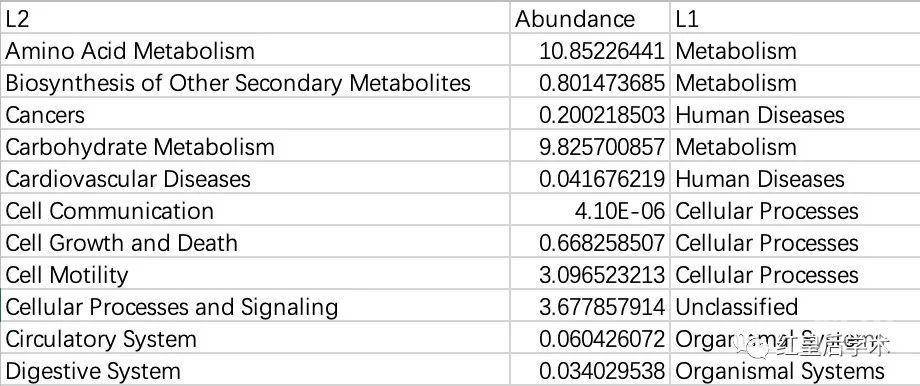
⚠️因为目的是获得所有样本中的主要功能,因此绘图数据需要提前计算一下每一个代谢路径在所有样本中的平均丰度。
图像绘制使用ggplot2的分面模式来展示不同代谢路径所属的第一层KEGG信息,应用条形图表示每一个代谢路径的相对丰度。
首先导入分析数据。
KEGG <- read.table("L2.txt",header = TRUE,sep = "\t")这里有一点需要说明,因为KEGG第一层分类中有几个分类的名字特别长,因此需要处理一下,使其能够实现自动换行。
library(ggplot2)library(reshape2)接下来就可以画图了。
p <- ggplot(KEGG,aes(Abundance,L2)) +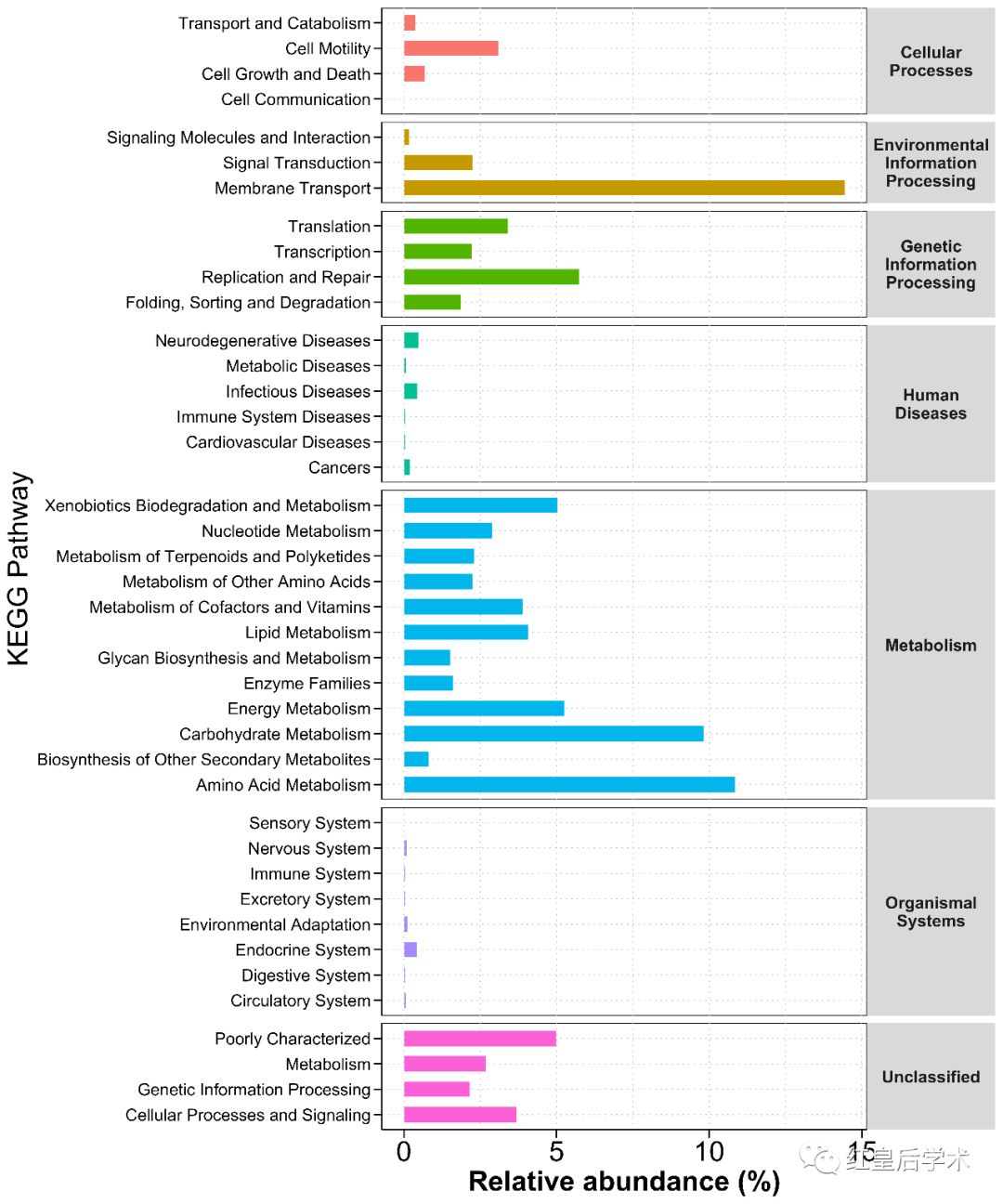
保存图片就OK了。
ggsave("L2.pdf", p, width = 183, height = 240, units = "mm")
png(filename="L2.png",res=600,height=7200,width=6000)PS:有些朋友可能想要把分面的文字放在图像的左侧,使其与代谢路径的名称放在一起,但是这个直接用ggplot2可能不行,因此把分面名称放在左侧之后,对其进行文字调整的参数就失效了,之前在群里也讨论过这个问题,据说是ggplot2的一个小bug,以后的更新中可能会修复。
来源:meta-genome 宏基因组
原文链接:https://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247491382&idx=2&sn=200f9eea6d2e84529d931f411246bab9&chksm=fab9f587cdce7c918d4b4ddc5351158b0b30ff6da7c501017e4f7d2bce7aa24ca3f02da5ad92#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
多特蒙德数据库
数据库

科技英才 | 周成虎:打造地理信息大数据库
文档数据库
“全球科研项目数据库(ProjectGate)”正式上线----中国科学院
空间环境信息数据库

汉语语言产生数据库发表
网状数据库

有人建了一个全球SCI撤稿信息数据库
「科情直通车」科技信息数据库建设遇难题


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号