科技工作者之家
科界APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-05-04
来源:CBG资讯
原标题:Nat. Catal.:Ni-Au双金属纳米粒子在CO2氢化过程中核壳结构的可逆损失
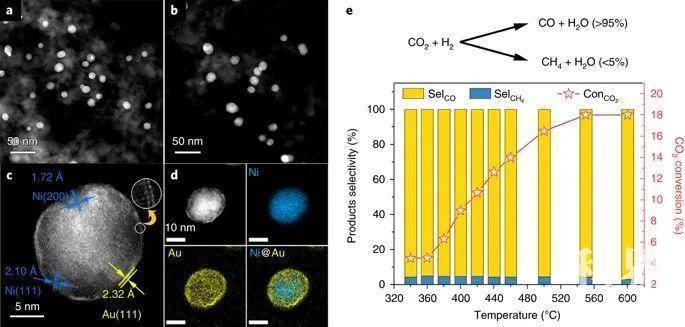 图1 NiAu/SiO2的微观结构和催化性能
图1 NiAu/SiO2的微观结构和催化性能(来源:Nature Catalysis)
Ni@Au/SiO2催化剂的CO2加氢催化性能测试 从图1e可以看出,CO2加氢反应的起始温度为340 ℃,转化率为~4.5%,在600 ℃时,转化率最高可达18%左右。CO是最主要的产物,在所有反应温度下的选择性均为95%。其他唯一的产物是CH4,这表明了与甲烷化反应(CO2 + 4H2 = CH4 + 2H2O)相比,该催化剂上的水气反向转移(CO2 + H2 = CO + H2O)是首选反应路径。 原位TEM观察反应过程中的表面重构动力学 通过瞬变电磁法(ETEM)观测可知,富Au原子的柱状结构显得较暗,而富Ni原子的柱状结构显得较亮。图2a显示了一个NiAu NP从450 ℃加热到600℃后,随后冷却至400 ℃的TEM图像,如图中黄色箭头所示。在450-500 ℃时,超薄的金壳在NiAu NP周围清晰地呈现为暗边缘。将温度提高到600 ℃后,暗边消失,说明最外层的Au族溶解在Ni基体中,形成了一种混合的NiAu合金。当催化剂冷却至450-400 ℃时,较暗的Au边缘重新出现,表明了Ni@Au核壳结构的恢复。研究者将原位成像限制在400 °C以内,仅能观察到Au壳的第一层偏析,这仍然证明了在CO2加氢反应条件下,Ni-Au体系合金过程的可逆性。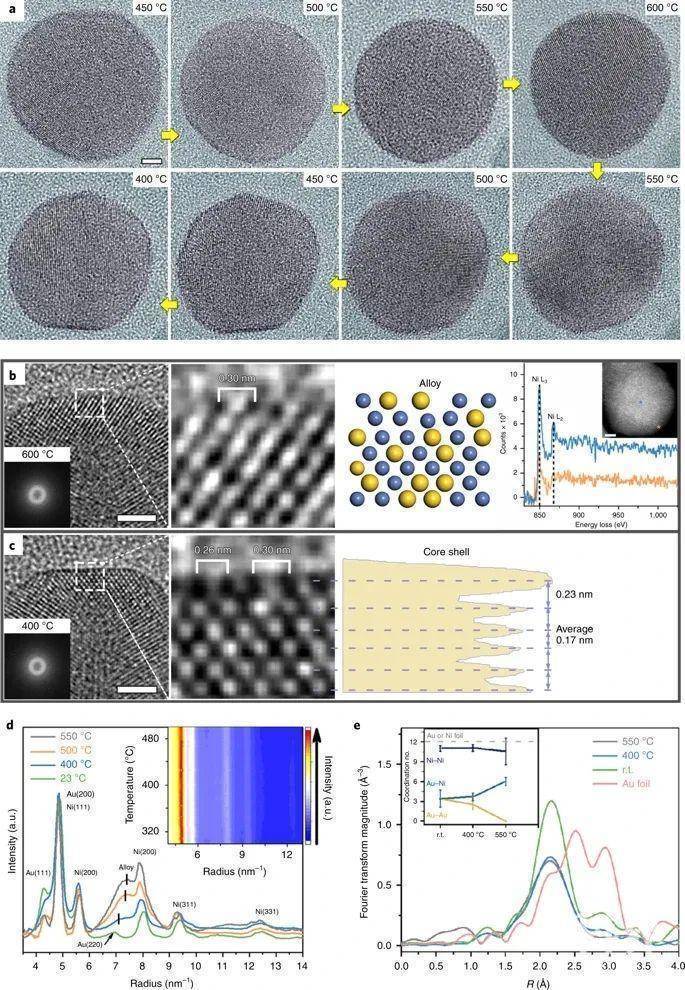 图2 反应过程中NiAu NP结构转变的原位观察
图2 反应过程中NiAu NP结构转变的原位观察 (来源:Nature Catalysis)
通过DFT计算分析了反应诱导重建机理 分离能(Eseg)被定义为单个Ni原子从块体到表面Au层所需的能量。如图3a所示,作者计算了不同反应物或中间体在CO2加氢过程中吸附和不吸附的Eseg。真空Eseg为1.31eV,表现出较强的Au表面偏析趋势,说明了初始Au壳在反应前和Ni@Au核壳结构恢复后的稳定性。CO的吸附导致了最大的还原,Eseg几乎为零,证实了Ni表面浓度的增加是由反应过程中CO的产生引起的。图3b的CO FTIR光谱显示了以CO为探测分子时,催化剂保留了富Ni的NiAu合金表面,但是在H2或N2氛围下仅出现富Au的表面。图3c中反映了重新分离的NiAu合金表面的反应活性和选择性,CO2在Ni表面位置被氢化时,最大的能垒为0.89 eV;而被吸附的CO分子从Ni位点扩散到Au位点时,能垒为1.23 eV,解吸为0.45eV。因此,可认为表面的Ni原子为CO2的氢化提供了活性位点,而表面的Au原子为CO的选择性生成提供了活性位点。
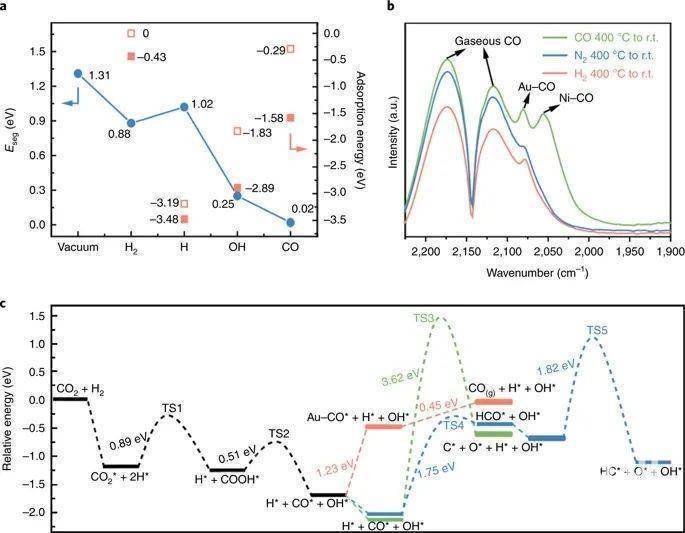 图3 NiAu结构演化的催化机制
图3 NiAu结构演化的催化机制来源:BeanGoNews CBG资讯
原文链接:https://mp.weixin.qq.com/s?__biz=MzI4ODQ0NjUwMg==&mid=2247500455&idx=2&sn=528f918db55d4aefd6d29d91be36bc24&chksm=ec3cc3eedb4b4af8ce763ab18bf75166afb156e8cf71d7ec3de6546f94963d40f0450a08d0ab#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

第二届微纳米马达会议

硅纳米粒子有助“曝光”肿瘤集聚区

Nat Comm | 在纳米粒子表面“拍”动画
山东大学奚宝娟:纸基碳支架负载核壳结构纳米粒子作夹层助力锂硫电池性能的提升

光伏纳米粒子可用作量子光源

“悬浮”纳米粒子助力量子计算

金纳米粒子新功能——探测缺陷

溯评单链聚合物纳米粒子
最新研究:纳米粒子“纠缠”突破量子极限

医用纳米粒子可为农作物输送营养


科界APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号