科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-07-21
来源:brainnews
ATP(Adenosine triphosphate, 腺嘌呤核苷三磷酸)最早是在1929年被Cyrus H. Fiske、Yellapragada Subbarow和Karl Lohmann分别发现。对于ATP的发现和认识过程是生命科学领域重要的研究进展,ATP相关的研究迄今为止已斩获4项诺贝尔奖。1972年,Geoffrey Burnstock在一篇发表在Pharmacological Reviews上的文章中首次提出了嘌呤能神经传递假说[1],认为ATP可以作为一种神经递质发挥功能。然而, 在接下来的二十年中,很多科学家都不支持甚至批判这一假说。这种质疑一定程度上是合理的,因为ATP在体内广泛分布,是生物体内各种生物化学过程的重要能源物质,让人怀疑其能否充当细胞外信使。这一争论直到20世纪90年代早期ATP和腺苷受体依次被克隆并表征,且1992年在交感神经节和大脑中发现了神经元-神经元突触传递后才得以平息,而嘌呤能假说也开始被科学家广泛接受[2]。
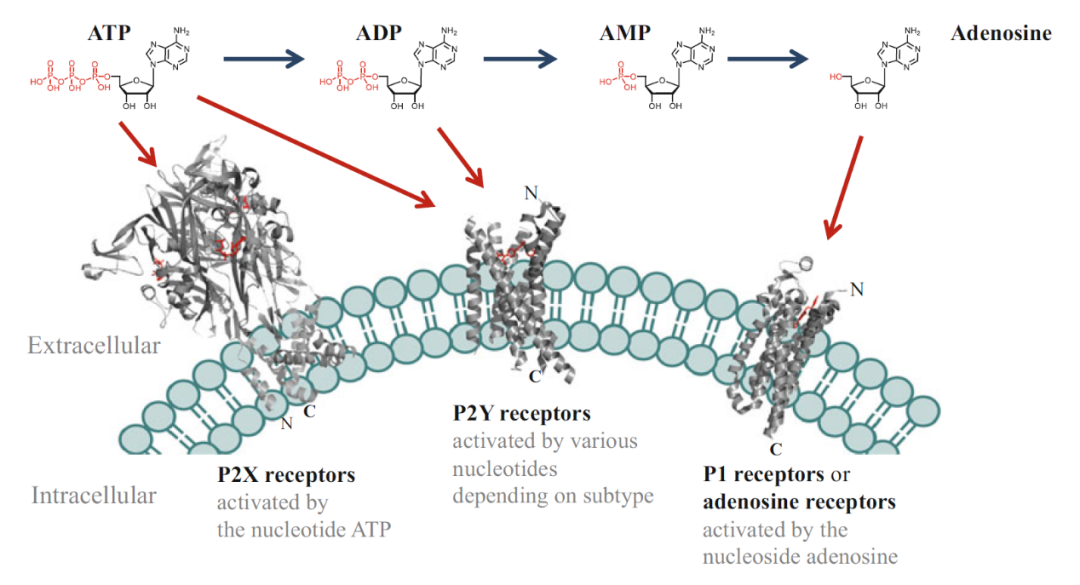
图一 嘌呤能受体分类[3]
1978年Geoffrey Burnstock提出“嘌呤能受体”(purinoceptor)。目前所发现的嘌呤能受体主要有以下几类(图一):P1受体(四个亚型,或称腺苷受体,A受体),其为G蛋白耦联受体,主要由ATP代谢物腺苷所激活;P2受体,分为P2X受体(七个亚型)与P2Y受体(八个亚型),能被ATP或其它核苷酸(如UTP)激活,其中P2X受体是一类配体门控离子通道,而P2Y受体为G蛋白耦联受体。P1受体与P2受体在嘌呤能神经传导中起到不同的作用,P1受体主要存在于神经肌肉接头前,介导了负反馈神经调节反应及递质释放的调节,而P2受体主要存在于神经肌肉接头后。除了在神经元细胞中的表达之外,嘌呤能受体也表达于多种非神经细胞上,其在突触传递、痛觉传导、炎症反应、心血管系统调节、免疫调节、肿瘤形成等多种生理和病理过程发挥作用的机制也引起了研究者的关注。
嘌呤能受体功能
嘌呤能受体已成为许多疾病的新型药物靶标[4]。在副交感神经中,ATP是乙酰胆碱的主要辅助传递因子,介导啮齿动物的膀胱收缩,但是在健康的人膀胱中,ATP作为辅助传递因子的作用很小。然而,在包括间质性膀胱炎,流出道阻塞和神经源性尿道膀胱功能障碍在内的病理状况下,嘌呤作为递质的成分增加至约40%。自发性高血压大鼠的交感神经中ATP的共递质作用也显著增强[5]。氯吡格雷和替卡格雷是抑制血小板聚集的P2Y12受体拮抗剂,是抗血栓形成和中风的非常成功的商业药物。类嘌呤化合物也正在开发中,用于治疗高血压和动脉粥样硬化,炎性肠病,干眼症,囊性纤维化,癌症以及许多其他疾病[6]。P2X3受体于1995年克隆,位于小颗粒(或小直径)伤害性感觉神经元上,存在于皮肤,舌头和内脏器官,有研究者提出了其引发疼痛的嘌呤能假说,以及描述内脏器官的嘌呤能机械感觉转导的假说。具体来说,扩张过程中上皮细胞释放的ATP作用于上皮下感觉神经末梢的P2X3和P2X2/3受体,从而产生痛觉。该假设的证据包括上皮释放ATP,P2X3受体在上皮下神经上的定位以及扩张过程中感觉神经中记录的反应,这些反应能够被外源性ATP模拟,并且P2X3受体拮抗剂可以降低这种反应。对P2X3基因敲除小鼠的研究表明,嘌呤能机械感觉传导在尿液排泄中也很活跃。对于神经性和炎性疼痛,研究者发现小胶质细胞上的P2X4,P2X7和P2Y12受体可能起到了关键作用,其拮抗剂在消除异常性疼痛方面非常有效。嘌呤能信号转导在创伤和局部缺血,包括阿尔茨海默氏症,帕金森氏症和亨廷顿氏病在内的神经退行性疾病以及多发性硬化症和肌萎缩性侧索硬化症以及神经精神疾病(包括抑郁症,焦虑症和精神分裂症)中的潜在作用也越来越受到研究者的关注[7, 8]。
2013年,南方医科大学高天明教授课题组发现ATP在星形胶质细胞调节成年小鼠抑郁样行为中起到重要作用[9]。他们观察到容易患上慢性社交衰竭障碍的小鼠大脑中的ATP含量较低,并且给予这些小鼠ATP能够诱导快速的抗抑郁样作用。缺乏肌醇1,4,5-三磷酸酯2型受体 (inositol 1,4,5-trisphosphate receptor type 2, IP3R2)和转基因阻断囊泡胶质传递 (gliotransmission),可诱导星形胶质细胞ATP释放缺陷,引起抑郁样行为,而给予ATP则可缓解这种行为。此外,他们还发现内侧前额叶皮层中的P2X2受体介导ATP的抗抑郁样作用。高天明教授课题组的这项研究揭示了抑郁症与嘌呤能信号转导相关的分子机制,从而为更深入地认识抑郁症病因机制,以嘌呤能受体为治疗抑郁症的新靶点的探究指明了新的方向[10]。作用于受体上的ATP多年以来都被认为是从神经元的胞吐小泡释放,或者是从受损或死亡的细胞中释放出来的。2018年,军事医学科学院基础医学研究所袁增强教授课题组揭示了星形胶质细胞可以通过CALHM2蛋白释放ATP[11]。结合其他实验,将CALHM2定义为ATP释放通道蛋白。Calhm2的常规敲除和条件性敲除均会导致ATP浓度显著降低,小鼠海马树突棘数量减少,神经功能障碍和抑郁样行为,而这些缺陷可以通过补充ATP得到明显缓解。2019年,浙江大学医学院沈啸教授研究组发表研究成果,系统分析了高血压下免疫系统的改变以及阐明了造成这一改变的始因[12]。该项研究提出了高血压下炎症的可测量指标,即血浆ATP浓度升高和抗原提呈细胞CD86的表达增强。这些指标有可能作为标记物被用来提示高血压相关病理进展严重程度。当然,血浆ATP浓度是否能够较血压提供更丰富的临床意义还需要进一步的临床试验来明确。其次,该项研究提示抑制红细胞ATP释放或者阻断P2X7受体有可能成为治疗高血压相关疾病的新策略。另外,临床上发现高血压与自身免疫病有很强的相关性,该项研究明确了ATP-P2X7-CD86通路的打开是介导这两类疾病相关性的分子机制之一。
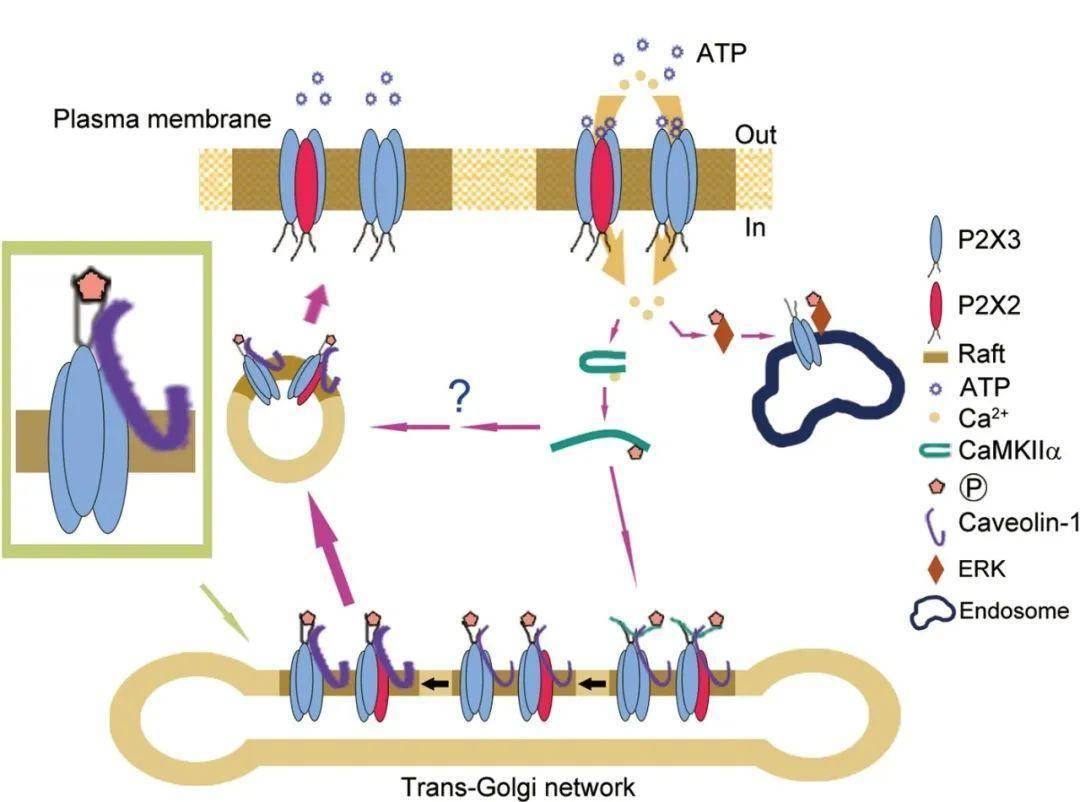
图二 P2X3 与P2X2/3受体上膜运输调控机制[14]
嘌呤能受体调控方式
基于嘌呤能受体的重要功能,对于嘌呤能受体的调控方式研究也受到了高度重视。2011年,中国科学院上海生命科学研究院生物化学与细胞生物学研究所鲍岚研究员研究组报道了P2X3受体新的信号传递机制:小GTP酶Rab5参与了P2X3受体进入内吞体的过程,Rab7则负责其长距离的逆向转运,P2X3受体的内吞和逆向转运都是受其配体ATP调控的,ATP激活的信号通路分子与内吞的P2X3受体一起进入到内吞体,形成了信号内吞体,神经元膜上的脂筏介导了P2X3受体的内吞和下游信号激活,信号内吞体进一步通过神经元轴突的逆向转运到胞体,调节胞体中转录因子CREB的磷酸化水平,同时影响神经元的兴奋性[13]。2014年,鲍岚研究员在上述工作基础上,更进一步发现受体的上膜运输受其配体的调控,需要 CaMKIIα和caveolin-1的协同作用,并可带动与其形成多聚体的P2X2受体的共转运 (图二)[14]。此项研究为痛觉传递中P2X3受体的功能调控提供了一种可能的机制。第二军医大学何成教授课题组则在2015年报道了Pirt蛋白通过抑制嘌呤能受体P2X3以调节膀胱过度活动[10]。Pirt蛋白是一种跨膜蛋白,其主要表达于周围神经元,但其在内脏中的生理和病理学作用尚不清楚。何成教授课题组发现缺乏Pirt蛋白的老鼠会导致膀胱过度活动,并且在Pirt蛋白缺陷型背根神经节神经元中,α,β-meATP诱导的电流密度显著增强。他们发现Pirt蛋白和P2X3受体共定位于膀胱神经纤维中,异源Pirt蛋白表达显著降低P2X3介导的电流。Pirt蛋白通过N末端的14个氨基酸残基与P2X3相互作用。偶联TAT的PirtN14肽可以抑制膀胱背根神经节神经元中的P2X3活化并减轻Pirt 缺乏小鼠的膀胱过度活动。在经环磷酰胺处理的小鼠的膀胱中,Pirt蛋白的表达降低,这是膀胱过度活动症的常用模型。重要的是,给小鼠使用PirtN14肽可减少其膀胱排尿的频率并恢复经环磷酰胺处理的小鼠的排尿体积[9]。
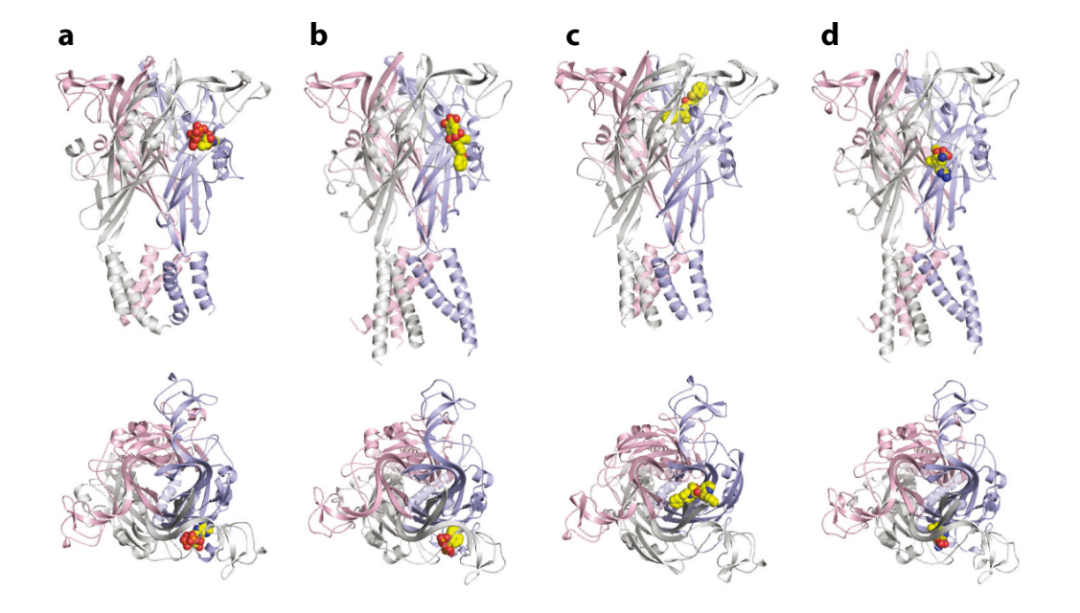
图三 P2X受体的激动剂和拮抗剂结合位点[22]
嘌呤能受体结构
对于嘌呤能受体结构的解析和变构机制研究有助于靶向嘌呤能受体的创新药物发现。最早解析出嘌呤能受体三维结构的是来自于美国俄勒冈健康与科学大学Eric Gouaux教授实验室,他们在2009年和2012年分别解析出了斑马鱼P2X4 (zfP2X4)受体静息态和开放态的晶体结构[15, 16]。与Eric Gouaux教授共同解析出zfP2X4开放态晶体结构的服部素之 (Motoyuki Hattori)教授目前任职于复旦大学。2018年,服部素之教授同当时在上海交通大学医学院工作的于烨教授团队一起揭示了首个P2X3受体的可成药性负性变构位点[17]。于烨教授一系列的工作还揭示了P2X受体的门控过程,特别是左鳍区域在门控过程中的重要性[18]。P2Y1R和P2Y12R是重要的抗血栓药物靶标。目前,己有四代靶向P2Y12R的药物上市,但这些药物存在副作用强、选择性差等缺陷。靶向P2Y1R的药物研发目前仍处于研究阶段,尚无药物成功上市,但某些药物先导化合物己体现出优于P2Y12R药物之处,例如缩短出血时间等,因此P2Y1R是一种非常有潜力的新型药物靶标。2014年,上海药物所赵强研究员和吴蓓丽研究员所领导的课题组成功测定了P2Y12R分别与激动剂和拮抗剂结合的复合物晶体结构,揭示了P2Y12R与不同药物分子的作用机制[19, 20];2015年,该研究团队进一步解析了 P2Y1R 分别与核苷酸类抑制剂 MRS2500和非核苷酸类抑制剂 BPTU 结合的复合物三维结构[21],这些结构的解析为新型抗血栓药物研发提供了新的依据。截止到目前为止,A1,A2A,P2Y1,P2Y12,P2X3,P2X4,P2X7等嘌呤能受体的结构已经得以解析(图三)。
靶向嘌呤能受体药物开发
自1972年以来,有关嘌呤能信号的论文有了惊人的增长,在Pubmed上,现在每年可以检索到接近2000篇相关的论文。尽管进行了数十年的研究,但迄今为止,与嘌呤受体相互作用的临床药物很少(表一),其中走在最前列的是P2Y12受体拮抗剂(氯吡格雷,普拉格雷,坎格雷洛和替卡格雷),它们已成为一类重要的抗血栓药物。另外瑞加德松(Lexiscan/regadenoson)是一种选择性腺苷受体激动剂,可使冠状动脉产生舒张,这款药物用于不能接受运动压力测试患者的放射性核素心肌灌注显像。然而紧随其后处于临床研发阶段的药物多达100-200个(数据来源参考AdisInsight)。其中就包括P2X3受体拮抗剂AF-219/gefapixant在慢性咳嗽和其他炎性疾病中的II期临床试验成功[23, 24],并在Merck 专利转让中产生了12.5 亿美元的交易额,目前正在多个国家同时开展III期临床实验。
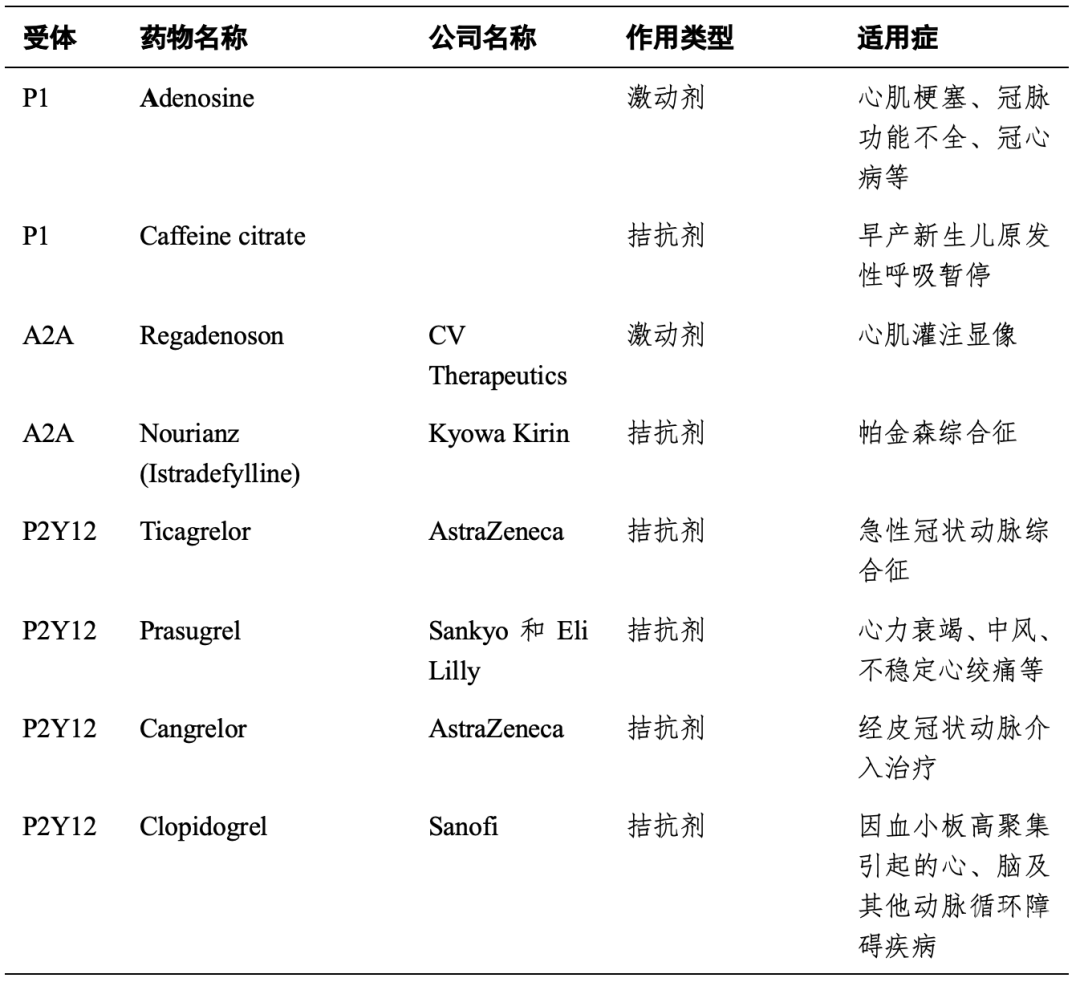
表一 以嘌呤能受体为靶点的临床药物
在受体功能研究与药理学中,“变构”(Allostery)一词出现的频率越来越高。1901年,研究者发现血红蛋白与氧气结合的协同作用存在变构现象, 1961年有研究者在冷泉港研讨会中正式使用变构的概念。自此,变构逐渐得以应用与发展。如今,变构调节已被认为是所有动态蛋白和其他生物相关大分子的固有特性,而以变构调节为基础的小分子药物则被称为变构调节剂。一般的蛋白质,包括酶、G蛋白耦联受体和配体门控离子通道,都可以被变构调节[25]。变构位点(allosteric sites),其与正构位点(orthosteric sites)不同,后者指的是内源性激动剂在受体上的结合位点。以P2X受体为例,除了通过与ATP结合位点(正构位点)相互作用来调节受体功能外,还可以通过与变构位点相互作用的分子来调节受体功能与结构。P2X受体ATP结合位点是由两个相邻亚基上五个带正电的碱性氨基酸和带芳香环侧链的氨基酸组成,这样导致作用于ATP结合位点的化合物(激动剂/拮抗剂)具有较高的极性,大多数情况下不符合成药的基本要求。因此,以P2X受体变构机制研究可以加快靶向P2X受体的创新分子发现,尤其在多个P2X受体亚型的晶体结构得以解析的“后结构时代”。实际上,上文所述的AF-219/Gefapixant即是通过变构调节来起作用的。
嘌呤能受体的功能在生物体内的几乎每个器官和系统中均扮演着重要角色。为特定的嘌呤能受体开发有效的激动剂或者拮抗剂是当下靶向嘌呤能受体药物研发的首要任务。我们应充分利用结构生物学的快速发展带来的“结构后时代”红利,充分利用受体结构、门控变化与变构机制等多种信息,提高高通量筛选效率与特异性,为定向的药物设计、合理的药物优化提供更多可能。相信在不久的将来,以嘌呤能受体为靶点的药物在临床运用中会越来越多。
来源:brainnews brainnews
原文链接:https://mp.weixin.qq.com/s?__biz=MzI2ODEyOTE3OQ==&mid=2649575224&idx=5&sn=001b40ca136f30bf408c3d27cb97d31e&chksm=f2eda9ccc59a20daed72eb023935413192b49b6d54965332eeaa005870b1a0f96f2ff8bf2856#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

火锅煮得好,嘌呤能变少?

发现第二种植物胞外ATP受体,揭示其在植物免疫中的作用

Cell:棕榈酰化如何抑制通道脱敏?——嘌呤受体P2X7全长结构解析
8-氮杂腺嘌呤
6-氯鸟嘌呤核苷

厦门大学林圣彩团队迎来开门红
6-糠氨基嘌呤
6-巯基嘌呤核苷
6-苄氨基嘌呤

Science | 首次揭示嘌呤小体的存在


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号