科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-09-14
来源:brainnews
小胶质细胞(microglia)是中枢神经系统(CNS)的组织驻留巨噬细胞,占成年哺乳动物大脑胶质细胞的5%-20%。小鼠中,小胶质细胞起源于胚胎卵黄囊中的红系-髓系前体细胞,迁移至胚胎脑内,随后自我更新维持数量。小胶质细胞从早期胚胎开始就与神经元和胶质细胞互作,在CNS的发育和稳态维持有重要作用。成年CNS中,神经元和先天免疫细胞的互作被认为与衰老和阿尔兹海默症(AD)等神经疾病有关联。事实上,被鉴定为AD进展常见风险因子的基因中,大部分或特异性表达于小胶质细胞,或在小胶质细胞中高表达【1】。而且,小胶质增生是神经系统疾病常见的组织学特征,包括小胶质细胞数量的增加以及小胶质细胞形态和转录的变化。由星形胶质细胞和小胶质细胞介导的慢性神经炎症是AD的病理特征之一。这些都提示小胶质细胞基因表达及其功能变化或参与疾病病理过程。
c/EBP家族是转录因子,调控小胶质细胞活化相关基因的表达,且在多种细胞因子和其他促炎基因的启动子和增强子区域有结合位点【2】,参与神经退行性疾病和脑损伤的炎症过程。c/EBPβ与人类的AD进展直接相关,其蛋白水平在AD病人的脑中显著上升。同时,老年AD小鼠模型的脑中Cebpb转录本显著上调。在小胶质细胞中表达c/EBPβ能提高神经毒性,LPS激活的野生型小胶质细胞诱导共培养的神经细胞死亡,而c/EBPβ缺陷的小胶质细胞不能,不过LPS激活的野生型小胶质细胞如何诱导周围神经元死亡还有待研究。减少Cebpb基因表达可逆转过表达tau小鼠的AD样病理学和认知障碍【3】。此外,c/EBPβ能促进小鼠实验性自身免疫性脑脊髓炎(EAE)的病理发生。小胶质细胞的失调参与多种神经退行性疾病,包括AD,但控制病理性小胶质细胞基因表达的机制目前还不清楚;c/EBPβ能促进小胶质细胞促炎基因的表达,在AD中被上调,两者之间是否存在调控关系以及c/EBPβ如何被调控都有待进一步的研究。
近日,来自Genentech公司的Kim Newton和Vishva M. Dixit在Cell杂志上发表文章Ubiquitin Ligase COP1 Suppresses Neuroinflammation by Degrading c/EBPb in Microglia,发现c/EBPβ在小胶质细胞中的表达受到泛素连接酶COP1的翻译后修饰调控。缺失COP1,c/EBPβ迅速积累,驱动促炎和神经退行性相关基因的表达,而缺失Cebpb的一个等位基因就能阻止促炎表型。缺失COP1同时增强小胶质细胞-神经元共培养的神经毒性,进一步研究发现该神经毒性几乎全部由补体介导。COP1缺失的小胶质细胞显著加速小鼠模型中tau介导的神经退行性。
研究人员首先发现Cebpb基因在巨噬细胞和小胶质细胞中高表达,但是c/EBPβ蛋白在没有刺激的条件下不能被检测到。使用蛋白酶体抑制剂MG132可导致野生型巨噬细胞中c/EBPβ蛋白快速增加,但不影响Cebpb mRNA,说明c/EBPβ蛋白经历了蛋白酶体降解。使用质谱检测,发现COP1是唯一能与内源的c/EBPβ共沉淀的泛素连接酶。COP1是多亚基culin-RING泛素连接酶CRL4COP1/DET1的底物适配器。COP1与DET1结合后,向DDB1-CUL4A-RBX1复合物传递蛋白质底物进行泛素化,适配的底物包括转录因子c-JUN、c/EBPα、ETV5等。共表达COP1和DET1显著降低c/EBPβ蛋白水平,突变COP1的底物结合位点则显著上调c/EBPβ蛋白水平。Cop1缺失导致小鼠胚胎第9天致死,所以采用可诱导性条件敲除。COP1敲除的原代小胶质细胞和骨髓来源的巨噬细胞(BMDMs)中,含有更多的c/EBPβ蛋白,敲除COP1互作蛋白STK40同样增加c/EBPβ蛋白,表明COP1在小胶质细胞和巨噬细胞中对c/EBPβ蛋白的翻译后抑制非常重要。LPS刺激1 h,小胶质细胞含有更多的c/EBPβ蛋白和COP1底物ETV5,即使是抑制了转录活性,说明LPS增加c/EBPβ蛋白含量不依赖于de novo(重新)基因转录。LPS激活MEK-ERK1/2激酶通路,随后抑制CRL4COP1/DET1,导致泛素连接酶的底物积累。利用LPS和ERK抑制剂显著降低细胞中c/EBPβ蛋白含量,表明ERK负向调控COP1依赖的c/EBPβ降解。
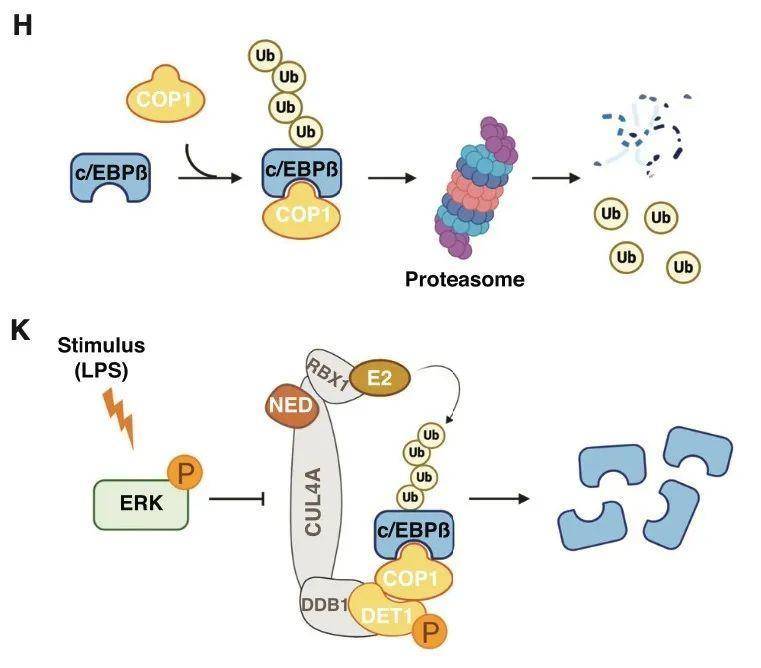
c/EBPβ调控参与小胶质细胞激活的基因表达,而COP1缺失导致其上调,那么COP1缺失是否改变小胶质细胞的基因表达。RNA-seq分析显示COP1缺失导致培养的小胶质细胞中参与炎症和宿主抵抗反应通路上调,与野生型小胶质细胞中,LPS诱导的基因表达类似;从小鼠中分离小胶质细胞进行RNA-seq分析,COP1缺失导致促炎基因如Ccl5、Cxcl2、Cxcl10、Il12b等,AD的主要风险因子Apoe,其他神经退行性相关的基因等显著上调。LPS相关、神经退行性相关、干扰素相关基因特征都在COP1 KO小胶质细胞中富集,而稳态相关基因则比WT表达较少。进一步利用单细胞RNA-seq对细胞的活化状态进行分析,WT和KO小胶质细胞共聚类成6个细胞亚群,其中COP1 KO分成具有神经退行性基因特征(61%)、具有干扰素相关特征(27%)两大类,而91%的WT则由稳态基因特征。即COP1的缺失促进小胶质细胞从稳态状态转换成激活状态,与神经退行性相关。
下一步,研究人员对COP1缺失条件下c/EBPβ如何直接靶向基因进行研究。在WT和COP1 KO原代小胶质细胞中进行c/EBPβ、H3K4me3(活跃启动子标志物)、H3K27ac(增强子区域标志物)的ChIP-seq分析。COP1 KO细胞中富集的c/EBPβ峰大多离转录起始位点较远,且H3K27ac信号增加;而WT细胞中富集的c/EBPβ峰大多在启动子附近。对COP1 KO特异性c/EBPβ峰的基因进行分析,发现与LPS响应、对细菌分子响应有关,说明c/EBPβ通过结合增强子区域调控促炎基因表达。
激活的小胶质细胞具有神经毒性。将小胶质细胞与神经元共培养,当WT小胶质细胞逐渐增加,神经元的死亡开始出现;然而,COP1 KO小胶质细胞显著增加神经元的死亡,敲除Cebpb的一个等位基因则能阻止COP1缺失导致的神经毒性和趋化因子和细胞因子增加。激活的小胶质细胞可通过补体途径导致神经元死亡,抗体阻断C1q可显著减少COP1 KO小胶质细胞导致的神经毒性,而中和TNF或IL-1R仅具有部分的保护作用。COP1 KO小胶质细胞含有更多完整的和切割的C3蛋白,分泌更多的C3,并且C3 mRNA水平增加,而缺失Cebpb的一个等位基因则能下调C3 mRNA水平。即COP1 KO小胶质细胞中,c/EBPβ驱动C3基因的表达增加。
最后,研究人员发现小胶质细胞特异性缺失COP1导致AD模型小鼠的激活型小胶质细胞和星形胶质细胞显著增加,神经退行性病变和海马萎缩更为严重。这种脑萎缩的增加与异常的Tau磷酸化水平增加、小胶质细胞增生、星形胶质细胞增加有关,这些都是神经炎症的标志。神经元标志物NeuN的免疫反应性较低,表示神经元丢失更加严重。因此,在tau介导的神经退行性病变小鼠模型中,COP1缺失加速疾病进展。
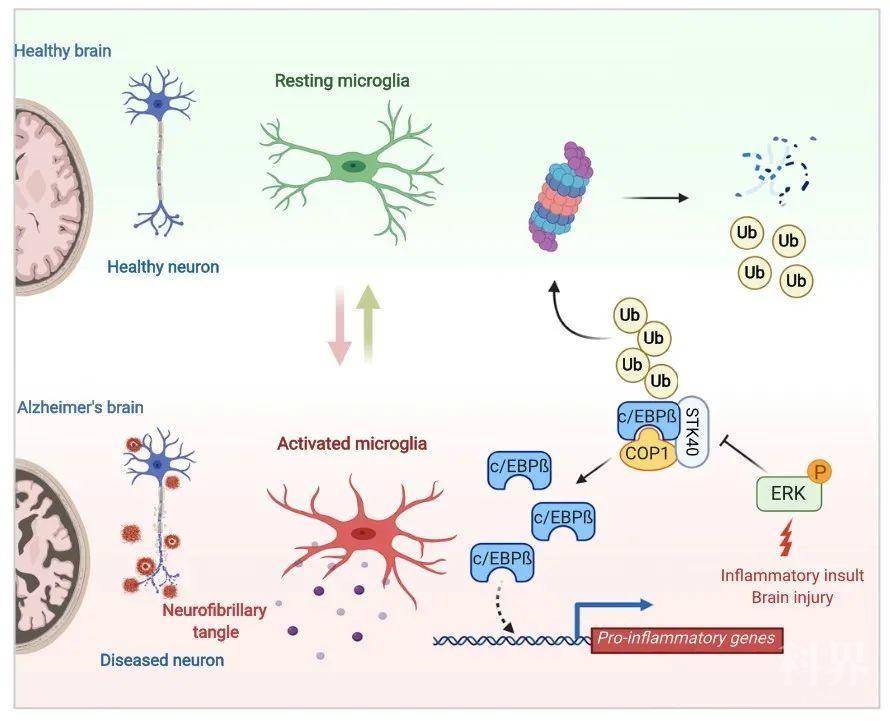
总的来说,文章揭示出COP1在翻译后调控小胶质细胞的c/EBPβ蛋白水平,c/EBPβ通过靶向增强子区域调控促炎基因的表达,进而影响小胶质细胞的激活状态。激活的小胶质细胞通过补体系统实现神经毒性,加快神经退行性病变。文章揭示了病理性小胶质细胞基因表达的新机制。
来源:brainnews brainnews
原文链接:https://mp.weixin.qq.com/s?__biz=MzI2ODEyOTE3OQ==&mid=2649577073&idx=5&sn=3b5447a76aa60563e726051cf50162fd&chksm=f2edb085c59a39935c069d1049245e44b9fffeaf7e562401e8f23303da90b895ca83f129ebb6#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
《自然》:小胶质细胞或能有效调节大脑中神经元的功能和行为!

夜班影响:基因表达不能适应新的睡眠模式

《自然》:把小胶质细胞和中间神经元放在一起养,探秘精神分裂症

从一篇10+文章看反义lncRNA如何调控基因表达

Cell:神经元释放的IL-33介导小胶质细胞促进突触重塑的神经机制

大型衰老相关基因表达谱数据库建成
小胶质细胞监测和保护神经元的功能
Ucp2依赖的小胶质细胞-神经元耦合调控焦虑样行为
大脑中的“接线员”:小胶质细胞塑造神经回路形成
研究揭示Cspg4high小胶质细胞在神经退行性疾病发病机制中的作用


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号