科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-09-04
来源:X一MOL资讯
八角属(Illicium)倍半萜华丽结构的人工合成,几十年来一直挑战着化学家的创造力。分离于大约40种植物,这些分子可根据其基本碳骨架进行分类,其中allo-cedrane、seco-prezizaane和anislactone家族的天然产物成员已有合成报道。Illisimonin A(1)是近来由中国医学科学院药物研究所的庾石山研究员、屈晶研究员的团队从Illicium simonsii 中分离而来,它是首例含有三环[5.2.1.01,6]癸烷碳骨架的倍半萜类化合物(图1)。Illisimonin A对缺氧缺糖诱导的SH-SY5Y细胞损伤具有神经保护作用,再者,目前从96 kg Illicium simonsii 果实中只分离到4 mg illisimonin A,原料的有限性限制了进一步的研究。

图1. Illisimonin A和其它八角属天然产物。图片来源:J. Am. Chem. Soc.
最近,加州大学欧文分校的Scott D. Rychnovsky教授课题组报道了(±)-illisimonin A的首例全合成,可一次制备94毫克外消旋产物。拆分中间体之后,可以合成(-)-illisimonin A,其绝对构型基于X射线结构分析数据修正为1S,4S,5S,6S,7R,9R,10R。
Illisimonin A的5-5-5-5-5五环骨架在合成上极具挑战,其有七个连续全取代的手性中心,其中的五个位于中心环戊烷环上,还包括两个相邻的季碳中心。此外,还存在一个反式戊烯环系。众所周知,反式5-5环系相对于顺式不稳定。除了Illisimonin A之外,此前仅有三个具有反式戊烯单元的天然产物被合成。充分考虑了分子内的张力、立体化学复杂性和空间拥挤程度,作者做出如下的逆合成分析(图2)。通过White酸介导的C-H键氧化来切开桥联内酯,在Maimone的假茴香素(pseudoanisatin)合成中有着相近的先例,官能团转换后逆推至2。C5-C6键通过semipinacol重排断开,经过常规转化后逆推至三环化合物3。3或类似结构中的两个环可以通过Diels−Alder反应来构建,逆推至5。最后,通过Aldol反应,5可以逆推至到6和7。中间体2具有1的全碳骨架,预计具有显著的环张力。通过在烃类9和8上MP2计算,作者估算了2的相对环张力及其可能的前体3,8具有约7.0 kcal/mol的能量优势,表明3的张力小于2。作者计划利用这一稳定性的差异,以分子内Diels-Alder反应首先合成4,其包含顺式5-5环。exo型IMDA(分子内Diels-Alder)反应的高选择性将引入4个手性中心,而在semipinacol反应中会出现张力更高的环系统,从而得到化合物2。环张力偏好增促进关键步骤IMDA反应的立体选择性。
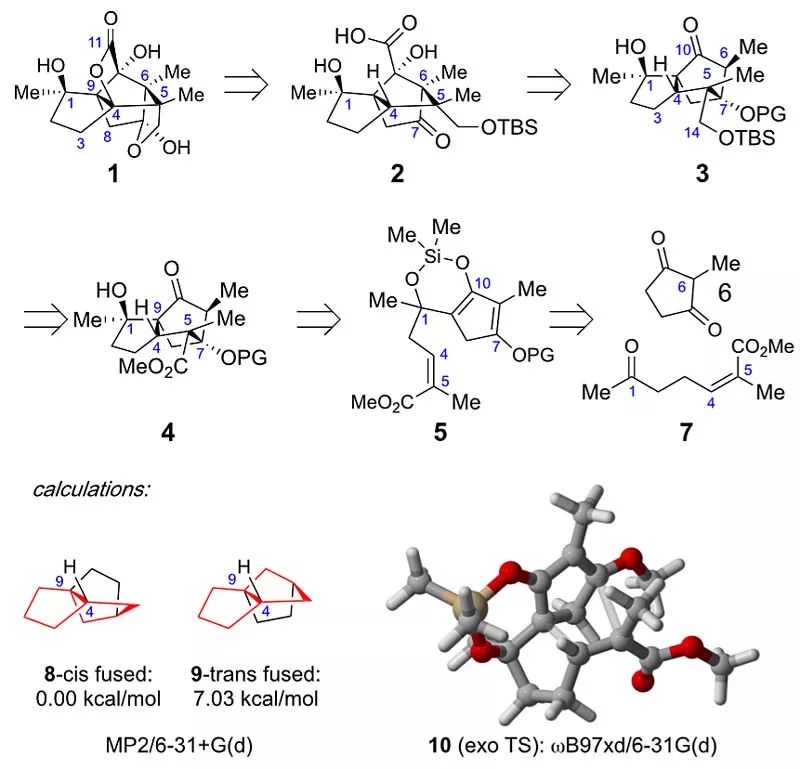
图2. 逆合成分析及环张力能量计算。图片来源:J. Am. Chem. Soc.
合成路线如图3所示。首先,2-甲基环戊-1,3-二酮(6)的烯醇形式以苄氧甲基醚的形式被捕获,产率优良。其次,与已知的酮7发生Aldol反应得到叔醇11,为非对映体混合物。在此阶段,作者从Bélanger课题组的研究中得到启发,用1,3-二氧-2-硅环己烯连接的化合物进行IMDA反应。硅环12由对映体混合物原位形成,加热至40 °C反应15 h,脱硅和纯化得到理想的Diels−Alder产物外消旋体13,为单一的非对映体,产率良好。据推测,叔醇的硅化导致环戊烯酮分子内活化,环戊烯酮在脱质子时被捕获为烯醇硅醚。合成的环戊二烯然后与亲双烯体发生Diels−Alder环化,其非对映面选择性由硅环模板控制。IMDA反应一步引入了五个手性中心和两个环。
在Diels-Alder反应中起着拉电子的作用后,13中的甲酯需要还原成伯醇。通过氢化铝锂全还原,TBS保护新形成的伯醇,再重新氧化C10酮,以良好的总收率得到14。14脱保护和由此产生的酮的溴化反应按预期得到溴化物19。如图3所示,在用银(I)处理后,未观察到预期的半缩醛产物。相反,分离得到ε-内酯21。很明显,C5的从C7到C6的semipinacol重排是为了产生所需的反式5,5-环系(20),但由此产生的半缩醛自发地经历了逆Claisen碎裂化。生成的20不适用于合成illisimonin A,为了避免这种反应,作者选择了在semipinacol重排之前引入C11碳。

图3. (±)-Illisimonin A的合成。图片来源:J. Am. Chem. Soc.
为了向C10酮中加入碳原子,作者尝试了许多种方法,最终用Barton的方法合成了乙烯基碘化物15。锂卤交换后进攻DMF并原位还原得到烯丙醇粗品,经m-CPBA氧化制得环氧16,两步收率83%。令人欣喜的是,在催化量的三氟乙酸存在下,在氯仿中进行3型semipinacol重排,克级规模也是成功的。竞争性的伯醇氢迁移是潜在的副反应,但在粗核磁谱中只观察到少量的醛。可见,相比σC11−H键,分子的刚性构象有利于σC5-C7键。在构建了illisimonin A中两个相邻的季碳中心和含有反式戊烯的基元后,再需要三步氧化就能得到目标分子。用标准化学方法将C11碳依次氧化为醛,然后再氧化为羧酸。用柱色谱法纯化α-羟基醛22时,重排成了α-羟基酮23。不使用柱层析,可避免重排。值得注意的是,illisimonane骨架可能是由eallo-cedrane骨架的碳阳离子重排而产生的。22到23的转化,是所提生物合成步骤的逆过程。
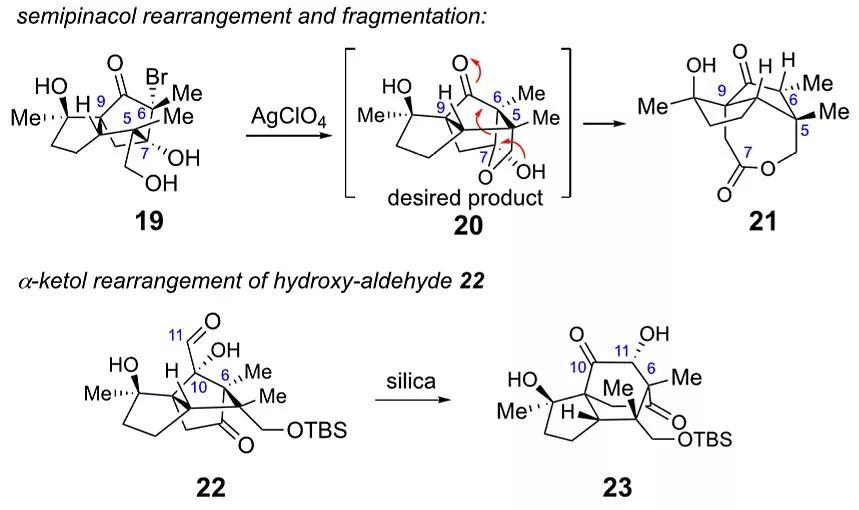
图4. 重排和碎裂化。图片来源:J. Am. Chem. Soc.
经过甲醇盐酸去保护后,只需一步C-H键氧化就能完成合成。由于C4次甲基是双新戊基位点,位阻极大,会阻止氧化。作者猜测,使用White的FePDP络合物催化剂催化,使用化学计量的过氧化氢作为氧化剂将酸18氧化为内酯1,其中六氟异丙醇(HFIP)的加入提高了18的溶解度,并通过氢键的电性去活化抑制了C14亚甲基的氧化副反应。18的刚性构象可能有助于C4次甲基的高反应活性。作者推测,在过渡态中张力释放也可能提高这个位置的反应活性。在最大规模的反应中,作者从196毫克的酸18得到了94毫克的天然产物1。
由于相关的Illicium天然产物在C1处有相反的构型,所以分离团队所确定的illisimonin A的绝对构型有些出乎意料。上文所述的外消旋合成并没有为讨论提供依据,但这是重新审视该问题的一个起点。在其它几种方法的基础上,以(S)-1-(1-萘基)乙基异氰酸酯为衍生剂,用柱色谱拆分了中间体烯丙醇(±)-24。对较低Rf非对映体26进行脱保护,转化为环氧(-)-16,并进一步衍生化。二茂铁羧酸(28)对C11醇的酯化引入了重原子。酯29的X-射线分析将26的绝对构型指定为1R系列。较高Rf非对映体27,推测具有1S构型,去保护并使用之前开发的方法得到(-)-illisimonin A。这种合成产物的CD与天然产物报道的一致。因此,天然产物(-)-illisimonin A的绝对构型为1S,4S,5S,6S,7R,9R,10R。

图5. 不对称合成及结构修正。图片来源:J. Am. Chem. Soc.
总结
加州大学欧文分校的Scott D. Rychnovsky教授课题组报道了(±)-illisimonin A的首次全合成。illisimonin A的核磁数据与天然产物相匹配,确定了天然产物的相对构型。对映选择性合成(-)-illisimonin A,结合X-射线结构,该课题组对天然产物的绝对构型进行了修正。合成路线中关键步骤包括1,3-二氧杂-2-硅杂环己烯模板化Diels-Alder环加成和3型semipinacol重排,最后步骤为铁催化的C-H键氧化反应。整条路线简洁高效,并可大量制备(±)-Illisimonin A,对探索其神经保护活性具有重要价值。
来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657618861&idx=2&sn=f6c273cb7182ebe68a527262c2cbbf12&chksm=80f8227db78fab6bf99c6ab06c2309ffc2a65dcf375c4d922c02c2a7612a99e2a089aa38280c&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

【纵览】一周科技新闻
遗传学:巨型基因组研究提供了关于人类疾病和遗传多样性的新知识

中国科技新闻学会倡议书

【纵览】一周科技新闻

2019年“湖南十大科技新闻”揭晓
神经科学:协同工作的大脑

【纵览】一周科技新闻

2023年国内十大科技新闻解读
2018年国内十大科技新闻解读
苹果WWDC 2017干货亮点全在这里


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号