科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-09-24
来源:化学加
导读
近期,诺丁汉大学化学系Ross M. Denton教授团队发展了一种催化Mitsunobu反应模式,其中通过一种全新的催化量氧化膦催化剂,代替了当量的三苯基膦和偶氮二羧酸酯,消除了氧化还原化学,消除了对末端氧化剂和还原剂的需求,并使得反应效率显著提高。文章发表在Science上,DOI:10.1126/science.aax3353。
跳转阅读→化工医药领域哪些上市公司、著名企业已经入驻化学加?
醇是化学合成中的重要原料,它们廉价易得,还可以通过亲核取代等方式转化成广泛的官能团。理想的亲核取代是羟基的直接立体定向取代,羟基构型翻转,同时生成唯一的副产物水(图1A),但实践中,动力学和热力学能垒阻止了直接取代,因此必须使用额外的化学活化剂对醇活化。常规方法如Mitsunobu反应(图1B)涉及当量的危险试剂且不符合原子经济性,产生了当量的三苯氧膦和肼衍生物。到目前为止,化学家们已经设计了多种策略来实现对π-活化的醇和亲核试剂的催化偶联,虽然有了一些进展,但要开发非活化的手性醇立体特异性的双分子取代仍然极具挑战。迄今为止,大部分工作都集中在通过当量试剂的氧化还原循环对传统Mitsunobu反应的改良上。但这种方法实现起来充满挑战,因为膦试剂的回收需要化学计量的还原剂,偶氮试剂的回收则需要互相兼容的化学计量的氧化剂(图1C)。
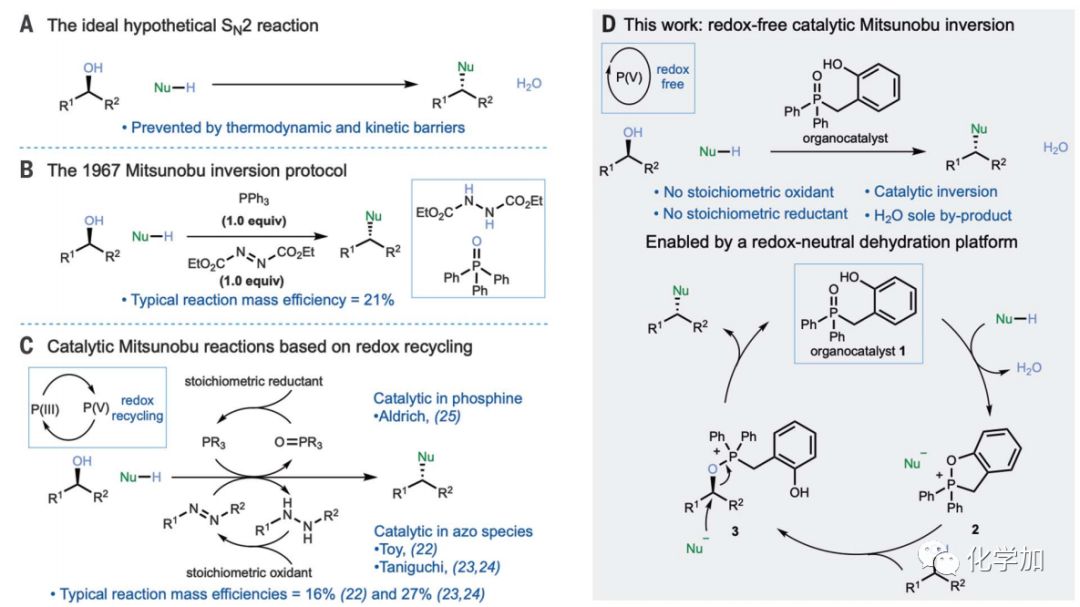
图1. 醇的双分子亲核取代反应(图片来源:Science)
催化的Mitsunobu反应最早报道于2006年,涉及2当量的三苯基膦和亚化学计量偶氮二羧酸酯 [10 mol%],偶氮二羧酸酯需用化学计量的氧化剂二(乙酰氧基)碘苯氧化回收。Taniguchi、Košmrlj等人在2013年和2016年的进一步工作中,使用的是一种改良的芳基偶氮羧酸酯,以分子氧为末端氧化剂,铁酞菁为共催化剂进行有氧氧化。这些过程成功地使Mitsunobu反应中氧化剂降低到催化量,但仍然需要化学计量的三苯基膦。在膦或两者都是催化量的反应中,产率较低。
近期,诺丁汉大学化学系Ross M. Denton教授团队设想是否可以开发一种替代催化模式,其中磷的氧化态不变,以催化的方式从磷(V)中产生Mitsunobu-活性磷化合物。因此,作者设计了一个基于氧化膦的催化剂1(图1D)的循环,作者推测它将被酸性前亲核试剂活化,并进行循环和脱水得到氧鏻盐2,通过醇开环得到常规中间体烷氧基鏻-亲核离子对3。随后发生亲核取代应得到产物并再生膦氧化物1,完成催化循环。这种方法没有氧化还原变化,水是唯一的副产物。
首先,作者研究了酸性前亲核试剂的作用,在假设的催化循环中,它参与了最初的脱水步骤。用催化剂1和(+)-2-辛醇进行实验,甲苯或者二甲苯为溶剂,使用Dean-Stark阱共沸除水对催化循环至关重要,因为鏻盐中间体是动力学和热力学不稳定的,易发生水解。低Brønsted酸性的亲核试剂如苯甲酸很难促进催化作用,但随着酸度的增加如4-硝基苯甲酸,则具有催化活性。这可能是脱水需要足够的质子活化强的磷-氧键。然而,随着酸度的增加,消除反应和酸促进的偶联,也变得越来越具有竞争力。最终发现二硝基苯甲酸可作为有效偶联配偶体用于构型翻转,并能以84%的产率和98%的ee值形成构型翻转的酯。
有了最佳反应条件,作者考察了催化Mitsunobu偶联反应的底物范围。如图2所示,含有潜在对强酸敏感基团的伯醇,包括酯(5a)、酰胺(5b)、邻苯二甲酰亚胺(5c)和腈(5d),都能在反应条件下进行酯化反应。β-香茅醇也得到了所需的酯产物(5e),其中敏感的三取代烯烃异构化较少。值得注意的是,含有膦敏感的烷基溴(5h)和叠氮化物(5i)的底物也能高效偶联,这在使用催化P(III)氧化还原循环策略时可能会出现问题。还可以使用对甲苯磺酸一水合物作为前亲核剂(5j),这提供了一种有价值的对甲苯磺酸酯的合成途径,避免了使用有毒的磺酰氯和化学计量比的碱。
Mitsunobu反应的标志是二级醇的构型翻转。无环、环状和苄基手性非外消旋仲醇与2,4-二硝基苯甲酸或2-硝基苯甲酸都能进行有效的反应。含有醚(5l),烯烃(5m),芳基氯(5o),砜(5q)和硅醚(5r)的底物都能以良好至极好的产率得到相应构型翻转的酯。当使用酸性较低的2-硝基苯甲酸作为偶联剂时,苄醇4t和4u得到所需的酯产物,具有优异的收率和翻转率。在较敏感的情况下,竞争性的消除会侵蚀产率,而立体化学完整性的损失可能是由于Fischer酯化或外消旋的一级亲核取代SN1反应所致。缺电子的醇也是挑战性底物,然而,对于醇4l,通过增加催化剂载量可以克服低反应性,得到5l,且具有优异的产率和选择性。天然5α-胆甾烷-3β-醇(4v)也能获得所需的酯。胆固醇(5x)和外型-降冰片醇(5w),也能形成相应的酯,由于底物本身的性质,使得构型保持。
接下来,作者尝试了使用该方法形成碳氮和碳硫键。使用二苯磺酰亚胺作为前亲核试剂,可以得到一系列N,N-双磺酰胺衍生物(5y到5ab)。N,N-双磺酰胺部分可以脱保护得到磺酰胺或伯胺。与(+)-2-辛醇的反应良好,(5ab)立体化学得到翻转。也可以通过使用硫代苯甲酸和1-癸醇作为偶联配偶体得到硫酯(5ac),证明碳硫键形成的可行性,但效率较低。
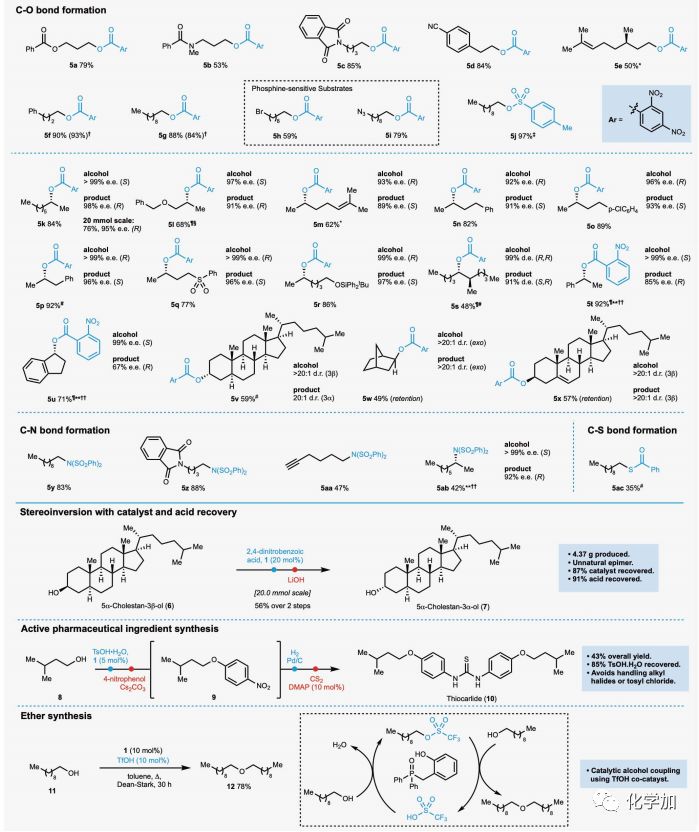
图2. 底物范围(图片来源:Science)
作者还采用催化Mitsunobu酯化法,再经酯水解,两步合成了克级规模的甾体5α-胆甾-3β-醇(7)的非天然异构体(4.37g,56%),两步间仅需溶剂交换。从最终混合物中回收催化剂1和羧酸,收率分别为91%和87%,用于底物5f和5g的催化酯化反应,产率无损失。
为了进一步验证新的Mitsunobu反应的范围和适用性,作者接下来研究了酚作为偶联配偶体的情况。虽然苯酚本身不足以作为前亲核试剂直接参与催化Mitsunobu偶联,但可以开发一锅法对甲苯磺酰化-酯化反应。以合成抗结核剂硫代卡利得10为例,异戊醇与一水合对甲苯磺酸反应生成异戊酸酯,并与4-硝基苯酚原位反应得到醚产物9。该一锅法醚化工艺避免了对甲苯磺酰氯或三溴化磷等有毒试剂的使用。随后还原和硫脲的生成得到了活性药物成分10。
最后,作者证明偶联的醇产物可以直接用作亲电试剂的反应模式。当三氟甲磺酸与膦氧化物1用作共催化剂时,Mitsunobu生成的烷基三氟甲磺酸酯足以与剩余的醇进行原位烷基化,得到对称的醚12,并再生三氟甲磺酸。该共催化模式避免了有毒物质的使用。
为了评估图1D所示的催化脱水反应,作者对2,4-二硝基苯甲酸和18O富集的1-癸醇的反应进行了研究(图3A)。结果表明,酯产物具有较高的16O掺入量,回收后的催化剂中18O含量为74%,这与预期的从醇到催化剂的氧转移是一致的。在没有催化剂的情况下进行了初步控制实验,反应30 h后,苯甲酰酯的产率为10%,构型保持产物的ee值为19%。作者推测,在反应过程中,立体化学完整性的丧失是由于Fischer酯化和消旋SN1机制的结合。接下来,作者探讨了膦氧化物13和14的作用,它们都没有催化活性(图3B)。在这两种情况下,可能的五元鏻物种2的形成都被阻止了。
作者接下来通过使用31P和1H谱监测反应来鉴定反应中间体。然而,在催化反应的等分试样中观察到的唯一磷物种是氧化膦1。由于活化的磷物种中间体通常对水解敏感并且氧化膦活化需要在Dean-Stark条件高温脱水,作者设计了一种替代方法来获得可能的催化中间体以避免产生水。为此,在室温下用三氟甲磺酸酐活化氧化膦1(图3C)得到了膦物种,其31P,13C和1H NMR数据与三氟甲磺酸鏻的一致,随后加入癸醇得到无环烷氧基三氟甲磺酸鏻3。最后,三氟甲磺酸鏻2被证明具有催化活性并且促进了膦氧化物1的合成(图3D)。
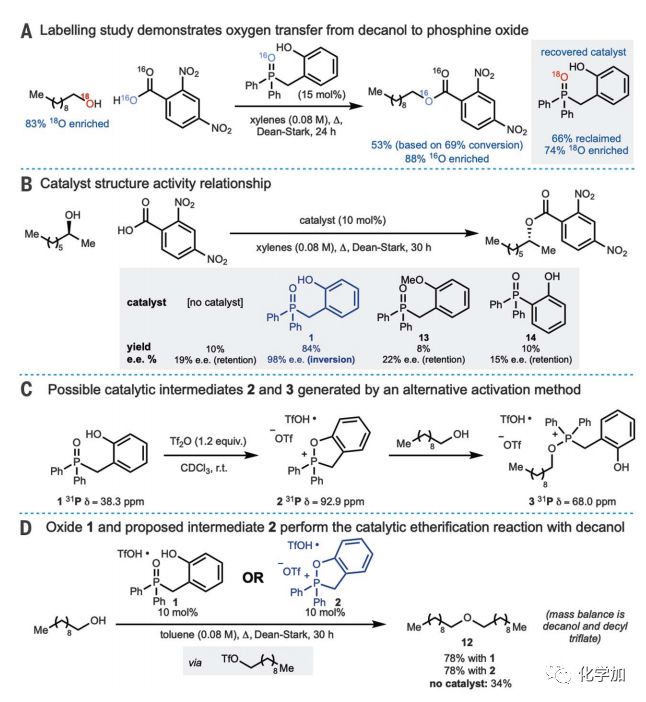
图3. 机理研究(图片来源:Science)
总结:诺丁汉大学化学系Ross M. Denton教授团队发展了一种催化Mitsunobu反应模式,其中通过一种全新的催化量氧化膦催化剂,代替了当量的三苯基膦和偶氮二羧酸酯,消除了氧化还原化学,消除了对末端氧化剂和还原剂的需求,并使得反应效率显著提高。这种有机膦催化的脱水模式在一系列其他经典的膦介导的转化中也具有潜在的应用价值。
来源:tryingchem 化学加
原文链接:https://mp.weixin.qq.com/s?__biz=MzAwODA5MjQ3Ng==&mid=2653086993&idx=1&sn=3f1141d248a2af094125cca21c21daae&chksm=80a26b42b7d5e254cd6b2d966ecba231659b7116c24a5bdc272debf94085b805f3792c299de7&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号