科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-09-26
来源:BioArt
20世纪以来,尽管人类对疾病生物学的认知和技术取得了显著的飞跃,但由于药物在临床试验阶段的高失败率,将新药推向市场仍然是一个耗时且昂贵的过程。计算机辅助小分子药物设计研究致力于以更低的市场成本研发新药。近年来,由于计算机数据处理能力的提高和新的人工智能(Artificial Intelligence,AI)方法的发展,人工智能逐渐进入药物设计科学家的视野。2020年8月,Journal of Medicinal Chemistry(JMC)推出 "Artificial Intelligence in Drug Discovery "专刊(https://pubs.acs.org/toc/jmcmar/63/16),聚焦于人工智能对药物发现过程的冲击,介绍了人工智能在药物研究中最新的方法和应用,让研究者了解到人工智能是如何进入药物发现领域的。特刊中的文章涵盖了药物发现的各个阶段:包含了药物分子的表示学习研究、机器学习模型的可解释性等理论研究;激酶抑制剂的发现,药物ADME/T(吸收、分布、代谢、排泄/毒性)性质预测等具体应用;还介绍了启发式药物发现推荐系统,药物分子的自动从头设计,药物合成路线设计等系统性的研发方法。这些内容展现了人工智能正在为药物研发提供广阔的新天地【1】。

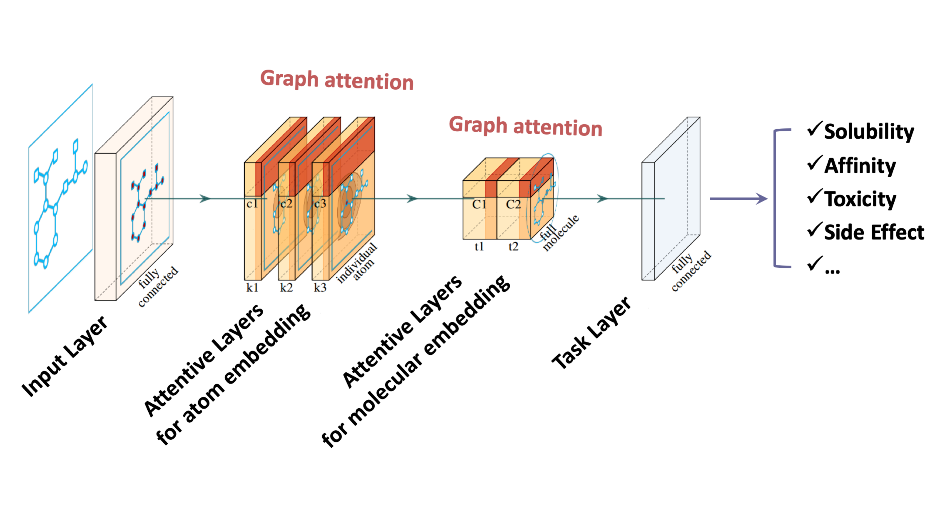
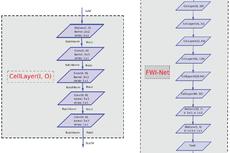
由郑明月和蒋华良领衔的中国科学院上海药物研究所和上海科技大学免疫化学研究所联合团队的两项研究成果入选这一专刊。其中,“Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism”【2】介绍了一种基于注意力机制的图神经网络模型(Attentive FP)(特别评述 | 人工智能助力药物研发:可解释性深度神经网络分子表征模型)。该模型可以用于分子表征,在多个药物发现相关的数据集上的预测表现达到当前最优,并且该模型所学到的内容具有可解释性。这种可解释性在机器的认知和人的认知的差异间架起了一座桥梁,由此可能更好地利用机器的认知增强药物学家的认知,产生更大的实际应用价值。Attentive FP的特征可视化表明,它可以自动从特定任务中学习到分子结构内非局部的特性,因此可以帮助药物学家或化学家超越经验和直觉,直接从各种性质数据中获取对该分子结构更深层的理解。
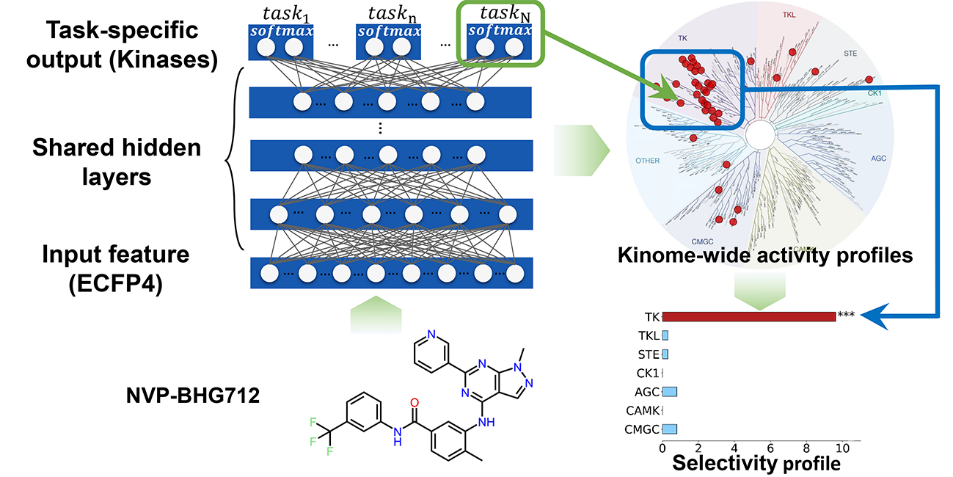
“Deep Learning Enhancing Kinome-Wide Polypharmacology Profiling: Model Construction and Experiment Validation”【3】描述了一种基于激酶活性大数据的药物激酶谱预测分析的多任务深度神经网络(multitask deep neural network)方法(Bioart报道链接)。多任务深度神经网络通过任务间的迁移学习,可以高效解决具有相关性的多类别分类问题。对于众多激酶靶点,共享的保守催化域使得多重活性预测任务紧密相关,因此利用多任务深度神经网络可以有效减少特定激酶靶点数据不足而无法建模的限制。该模型在与同类型方法的平行比较中显示了更好的预测效果,并对多个大规模外部测试集展现了良好的泛化性能。此外,该研究团队使用模型预测了数个临床或在研的激酶抑制剂,并进行了生物实验验证。结果证明模型不仅可以准确识别抑制剂已知的主要靶标,还可以推断出与毒副作用相关靶标的潜在作用,对多靶点药物分子的精准设计具有重要指导意义。
原文链接
https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000860
参考文献
1.Bajorath, J., etal., Artificial Intelligence in Drug Discovery: Into the Great Wide Open.Journal of Medicinal Chemistry, 2020. 63(16): p. 8651-8652.
2.Xiong, Z., et al.,Pushing the Boundaries of Molecular Representation for Drug Discovery with theGraph Attention Mechanism. Journal of Medicinal Chemistry, 2020. 63(16): p.8749-8760.
3.Li, X., et al.,Deep Learning Enhancing Kinome-Wide Polypharmacology Profiling: ModelConstruction and Experiment Validation. Journal of Medicinal Chemistry, 2020.63(16): p. 8723-8737.
来源:BioGossip BioArt
原文链接:https://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652501143&idx=6&sn=74ec7c40d7e1556f72180a214ad90c62&chksm=84e26723b395ee35bd334f6ed458c75717fd7310711b9323dcaa530a739a1d8cc2c7e24a5682#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

出版简讯 | 人工智能与大数据助力靶向药物研发

人工智能技术有望提升药物研发效率
人工智能 | 500个闪光的“浮动代码”,魔性的人工神经网络

人工智能使神经网络设计速度提高200倍
类脑量子叠加脉冲神经网络:从量子大脑假说到更好的人工智能
“传播科学火种·启迪科学梦想”科普志愿新疆行举办




人工智能或改变药物研发游戏规则
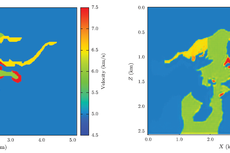
Geophysics:人工智能深度神经网络地震速度分析技术
自治区科协举行“院士专家进校园”科普报告会


何清龙等-Geophysics:人工智能深度神经网络地震速度分析技术



科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号