科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-10-25
来源:BioArt
大规模人群的全基因组测序研究有助于理解人类演化和迁徙的历史,以及发现与疾病相关的遗传变异。人类的遗传变异绝大部分为罕见变异,只出现在特定人群中。例如,国际千人基因组项目发现的8千多万个单核苷酸多态性(Single Nucleotide Polymorphism, SNP)位点中,大约90%的最小等位基因频率(Minor Allele Frequency, MAF)小于0.05,且86%仅出现在单一大陆人群中【1】。
然而,现有人类基因组学研究以及公共数据库的样本绝大部分来自欧美人群,所取得的研究成果并不完全适用于非欧美人群,严重限制了精准医学的实施和推广。亚洲人口占世界总人口的60%,具有丰富的遗传多样性。新加坡由于其独特的近代移民史,形成了主要包括华人、马来人和印度人的人口组成,是亚洲人群遗传多样性的一个绝佳缩影,因此针对新加坡人的全基因组测序研究将推进我们对亚洲人群特有的基因变异的了解,对精准医学在亚洲的发展具有重要意义。
2019年10月17日,新加坡万人基因组联盟(SG10K Consortium)在Cell上发表了名为“Large-Scale Whole-Genome Sequencing of Three Diverse Asian Populations in Singapore”的封面文章,报道了对2780个新加坡华人,903个马来人,1127个印度人的全基因组测序数据的群体遗传学研究成果。

此研究采取13.7X的中深度全基因组测序设计,结合前沿生物信息学方法以及严格质控,发现了89160286个SNPs和9113420个插入缺失变异(INDELs),其中一半以上为尚未在国际遗传数据库dbSNP记录的新发现变异,从侧面反映了现有数据库中亚洲人群数据的严重匮乏。该研究还发现,有126个新发现变异被生物信息软件(Polyphen和SIFT)预测为有害,但这些变异在新加坡人群中却是MAF>0.01的常见变异。罕见遗传疾病(rare diseases)基因诊断的一个重要步骤是通过对比公共数据库中报道的等位基因频率对候选致病基因突变进行筛选,上述结果提示基于现有公共数据库对亚洲人进行疾病基因诊断的准确性将受到影响,限制了精准医学的发展。
该研究还进行了深入的群体遗传学分析,系统刻画了三大亚洲族群的人口学特征和历史。作者通过估计每对个体的亲缘系数,发现近亲(second cousins以内)婚配在新加坡印度人、马来人和华人中的流行率分别为29.1%,10.8%和2.6%。这个数据对公共卫生有重大意义,因为近亲结婚将导致子代携带更多的致病基因纯合突变,增加早夭或患隐性遗传病的概率。另外,作者通过与千人基因组项目中10个亚洲人群的数据进行比较分析,证实了新加坡三大族群涵盖了广泛的亚洲人群遗传多样性,且每个族群内部还有更精细的群体遗传结构。作者估计大约96%的新加坡华人来自中国南方,大约4%来自北方,而大约81%的新加坡印度人来自印度次大陆的南方(包括印度南部、孟加拉国、斯里兰卡等),大约19%来自北方(包括印度北部、巴基斯坦等)。而新加坡的马来人则具有东南亚原住民的遗传学特征,却并未被千人基因组项目报道。因此,作者重点对马来人进行了更深入的人口学历史推断,发现代马来人和华人的共同祖先在约45,900年前与印度人分离,而马来人和华人则在24,800年前分离。另外,马来人的祖先大约在1700(95%置信区间1469-1890)年前与来自东亚的人群有过显著的基因混合(admixture),其中来自东亚的人群的遗传特征可在现代的越南京族人,中国南方汉人,以及傣族人中发现。通过遗传学数据推断的马来人基因混合时间以及混合人群特征均与考古学发现的源自台湾的南岛原住民扩张历史(Austronesian expansion)吻合。这些研究发现加深了我们对亚洲人群遗传结构和演化历史的认识。
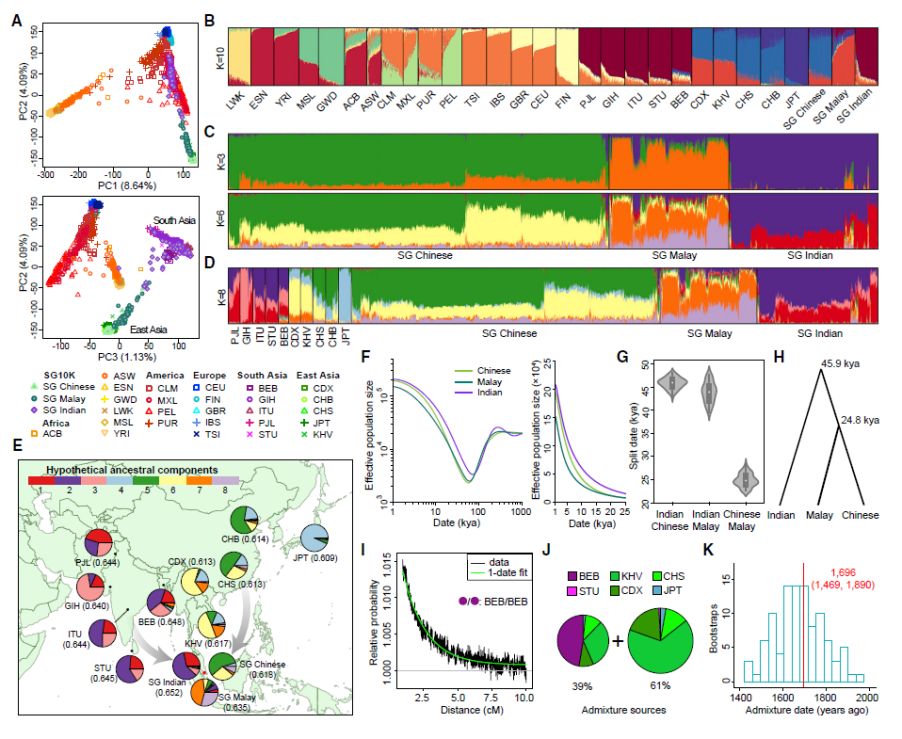
另外,作者还通过对比三大亚洲族群的等位基因频率,使用Population Branch Statistics (PBS)对基因组进行扫描,发现了20个可能受到自然选择的基因位点。其中,7个位点在之前的研究中被广泛认可在亚洲人群中受到自然选择作用,包含的基因有EDAR与PRSS53(与毛发形态有关), OCA2(与浅色皮肤有关), ALDH2与ADH1B(与酒精代谢有关), HYAL2(与细胞对紫外线UV-B的应激反应有关)和IL4(与对病原体的免疫反应有关)。其余13个位点为新发现位点,其中信号最强的有FN1(与冠状动脉疾病有关),FAM178B(与精神分裂症有关)和CENPW(与代谢有关)。作者还系统检索了已报道的位于这20个位点中的全基因组关联研究(GWAS)信号,旨在探索自然选择对人类疾病和相关性状的影响。他们发现GWAS 信号在受自然选择的位点里面有显著的富集效应,20个位点中有14个包含了已报道的GWAS信号,其中7个位点里面自然选择的index SNP与GWAS SNP存在连锁不平衡,揭示了自然选择对人类表型多样性的形成具有重大影响。
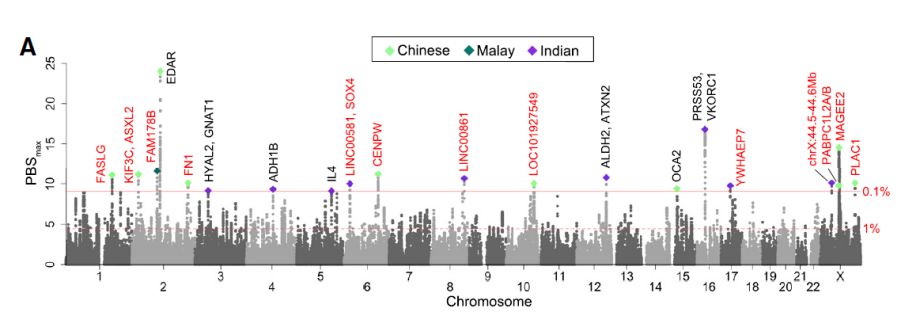
最后,作者通过基因型填补(genotype imputation)实验来检验新加坡人群数据对推动亚洲人群遗传学研究的作用。作者以来自世界各地56个人群的GWAS芯片基因分型数据作为填补对象,比较了传统的基于千人基因组测序数据的填补方法和加入了新加坡SG10K数据的填补方法。作者发现,得益于新加坡人群丰富的遗传多样性,结合千人基因组测序数据和SG10K可以显著地提高绝大部分亚洲人群和大亚洲人群的基因型填补精度,尤其是对于罕见变异的填补。例如,相比于只用千人基因组数据进行填补的方法,加入SG10K数据可将华人和马来人的填补错误率降低50%,将印度人的填补错误率降低22%。基因填补作为一项重要的生物信息学方法已经被广泛地应用于GWAS,而降低基因填补错误率将有助于增强GWAS的统计效能,对加速发现遗传疾病和性状的相关基因和药物靶点具有重要的意义。因此,这项研究所产生的数据和结果有望成为推进亚洲人群遗传疾病研究和精准医学发展的一项重要资源。
据悉,华中科技大学公共卫生学院王超龙教授和新加坡基因组研究所的刘建军教授为该文的共同通讯作者,王超龙教授课题组成员巫德刚,窦锦壮和柴晓冉为共同第一作者。
附:王超龙课题组招聘博士后
华中科技大学公共卫生学院王超龙教授课题组公开招聘博士后2名,诚邀热爱生物信息学与基因组学工作的科研人员加盟。
原文链接:
https://doi.org/10.1016/j.cell.2019.09.019
参考文献
1. 1000 Genomes Project Consortium, Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A., and Abecasis, G.R. (2015). A global reference for human genetic variation. Nature 526, 68–74.
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652476774&idx=1&sn=cbe3fbcfda3b82f212d8de9d71c44742&chksm=84e206d2b3958fc4f77a47d47fc69e287338dfd1b613af1e78adf1db0eb2524414d9ea22c3d9&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
基因组 测序深度 测序中国 全基因组测序 刘建军 全基因组关联分析 新加坡华人 马来人

中国“绿色超级稻”新品种累计推广面积9000万亩

ChinaMAP发布最大规模中国人群高深度全基因组测序和表型研究成果
ChinaMAP发布最大规模中国人群高深度全基因组测序和表型研究成果

日本完成一种毒蛇全基因组测序
全基因组关联分析学习资料(GWAS tutorial)

小麦D基因组完整图谱首绘成功

中国研究团队破解中国种茶树全基因组密码

首次对喀斯特植物实施全基因组测序

日本科研人员发现无呼吸微生物

动物全物种基因组测序“顾此失彼”


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号