科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-10-25
来源:宏基因组
摘要
宏基因组测序结果有应用于微生物检测和鉴定的潜力,但需要新的工具来提高其敏感性。在这里,我们提出了一种计算方法——CATCH,以增强核酸捕获丰富的各种微生物类群。CATCH可设计具有指定数量的寡核苷酸的最佳探针集,可实现已知序列多样性的完全覆盖和扩展。我们致力于在复杂的宏基因组样本中应用CATCH来捕获病毒基因组。我们设计、合成和验证多个探针集,包括一个针对356种已知感染人类病毒全基因组的探针集。用这些探针集捕获的病毒平均含量增加了18倍,这使得我们能够组装那些不浓缩就无法恢复的基因组,并准确地保存在样本多样性中。我们还使用这些探针组恢复2018年尼日利亚拉沙热爆发的基因组,并改进人类和蚊子样本中未鉴定病毒感染的检测。结果表明,CATCH可以实现更敏感和更经济有效的宏基因组测序。
Metagenomic sequencing has the potential to transform microbial detection and characterization, but new tools are needed to improve its sensitivity. Here we present CATCH, a computational method to enhance nucleic acid capture for enrichment of diverse microbial taxa. CATCH designs optimal probe sets, with a specified number of oligonucleotides, that achieve full coverage of, and scale well with, known sequence diversity. We focus on applying CATCH to capture viral genomes in complex metagenomic samples. We design, synthesize, and validate multiple probe sets, including one that targets the whole genomes of the 356 viral species known to infect humans. Capture with these probe sets enriches unique viral content on average 18-fold, allowing us to assemble genomes that could not be recovered without enrichment, and accurately preserves within-sample diversity. We also use these probe sets to recover genomes from the 2018 Lassa fever outbreak in Nigeria and to improve detection of uncharacterized viral infections in human and mosquito samples. The results demonstrate that CATCH enables more sensitive and cost-effective metagenomic sequencing.
图1. 使用CATCH设计探针组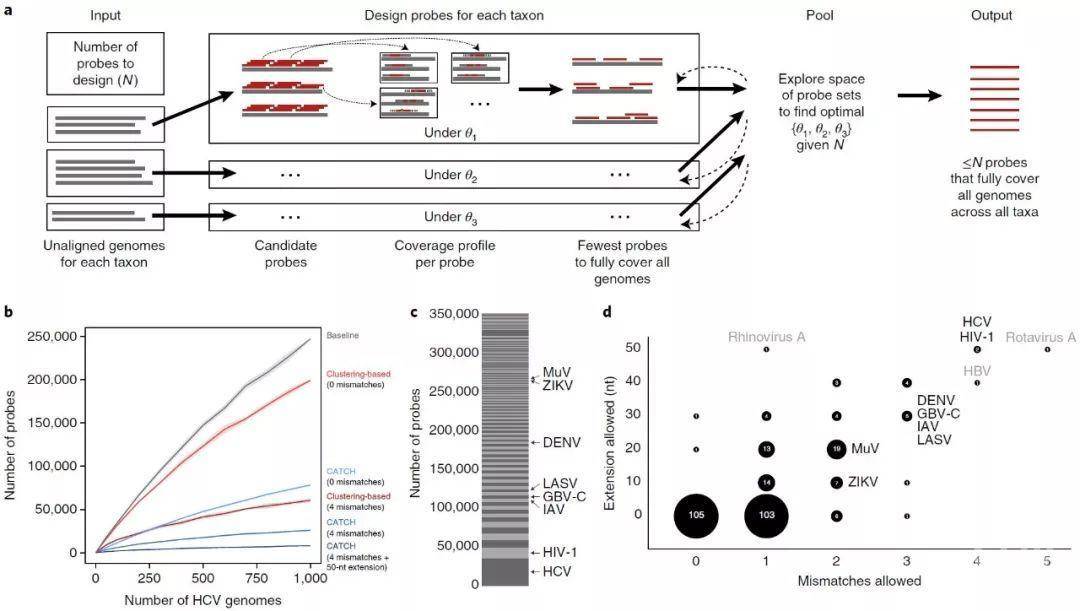
a,CATCH探针设计方法的概述,显示了三个数据集(通常,每个数据集都是一个分类单元)。对于每个数据集d,CATCH通过跨输入基因组平铺(tiling)来生成候选探针,并且可以选择使用位置敏感散列来减少候选探针的数量。然后确定每个候选探针在参数为θd的模型下杂交(基因组和其中的区域)的位置(详见补充图1b)。使用这些覆盖率曲线近似于完全捕获所有输入基因组的最小探针集合(在文本中描述为s(d,θd))。考虑到探针总数(n)的限制和θd上的损失函数,它搜索d所有的最佳θd.
b,完全捕获不断增加的HCV基因组所需的探针数量。所示的方法是简单的平铺(灰色),一种基于聚类的方法,在两个严格级别(红色)上,并使用三个参数值选择捕获,这些参数值指定不同的严格级别(蓝色)。参数选择详见补充说明2。以前针对病毒多样性的方法在探针集设计中使用聚类。每一行周围的阴影区域是随机抽样输入基因组计算的95%点置信区间。
c,CATCH为VALL探针集所有349,998个探针中的每个数据集(共296个数据集)设计的探针数。我们的样本测试中包含的物种都有标签。
d,CATCH为VALL设计中的每个数据集选择的两个参数值:假设在杂交中允许不匹配数量和杂交区域每侧的目标片段长度(以核苷酸为单位)。每个气泡的标签和大小指示分配给特定值组合的数据集数量。样本测试中包含的物种用黑色标记,未包含在测试中的异常物种用灰色标记。一般来说,多样性更高的病毒(例如,HCV和HIV-1)被分配的参数值(这里是高值)比多样性低的病毒更宽松,但在设计中仍然需要相对大量的探针来覆盖已知的多样性(见C)。用于设计VWAFR探针集时,类似于c和d的图在补充图3中。
Fig. 1 | Using CATCH for probe set design.
a, Sketch of CATCH’s approach to probe design, shown with three datasets (typically, each is a taxon). For each dataset d, CATCH generates candidate probes by tiling across input genomes and, optionally, reduces the number of them using locality-sensitive hashing. Then it determines a profile of where each candidate probe will hybridize (the genomes and regions within them) under a model with parameters θd (see Supplementary Fig. 1b for details). Using these coverage profiles, it approximates the smallest collection of probes that fully captures all input genomes (described in the text as s(d, θd)). Given a constraint on the total number of probes (N) and a loss function over θd, it searches for the optimal θd for all d.
b, Number of probes required to fully capture increasing numbers of HCV genomes. Approaches shown are simple tiling (gray), a clustering-based approach at two levels of stringency (red), and CATCH with three choices of parameter values specifying varying levels of stringency (blue). See Supplementary Note 2 for details regarding parameter choices. Previous approaches for targeting viral diversity use clustering in probe set design. The shaded regions around each line are 95% pointwise confidence bands calculated across randomly sampled input genomes.
c, Number of probes designed by CATCH for each dataset (of 296 datasets in total) among all 349,998 probes in the VALL probe set. Species incorporated in our sample testing are labeled.
d, Values of the two parameters selected by CATCH for each dataset in the design of VALL: number of mismatches to tolerate in hybridization and length of the target fragment (in nucleotides) on each side of the hybridized region assumed to be captured along with the hybridized region (cover extension). The label and size of each bubble indicate the number of datasets that were assigned a particular combination of values. Species included in our sample testing are labeled in black, and outlier species not included in our testing are in gray. In general, more diverse viruses (for example, HCV and HIV-1) are assigned more relaxed parameter values (here, high values) than less diverse viruses, but still require a relatively large number of probes in the design to cover known diversity (see c). Panels similar to c and d for the design of VWAFR are in Supplementary Fig. 3.
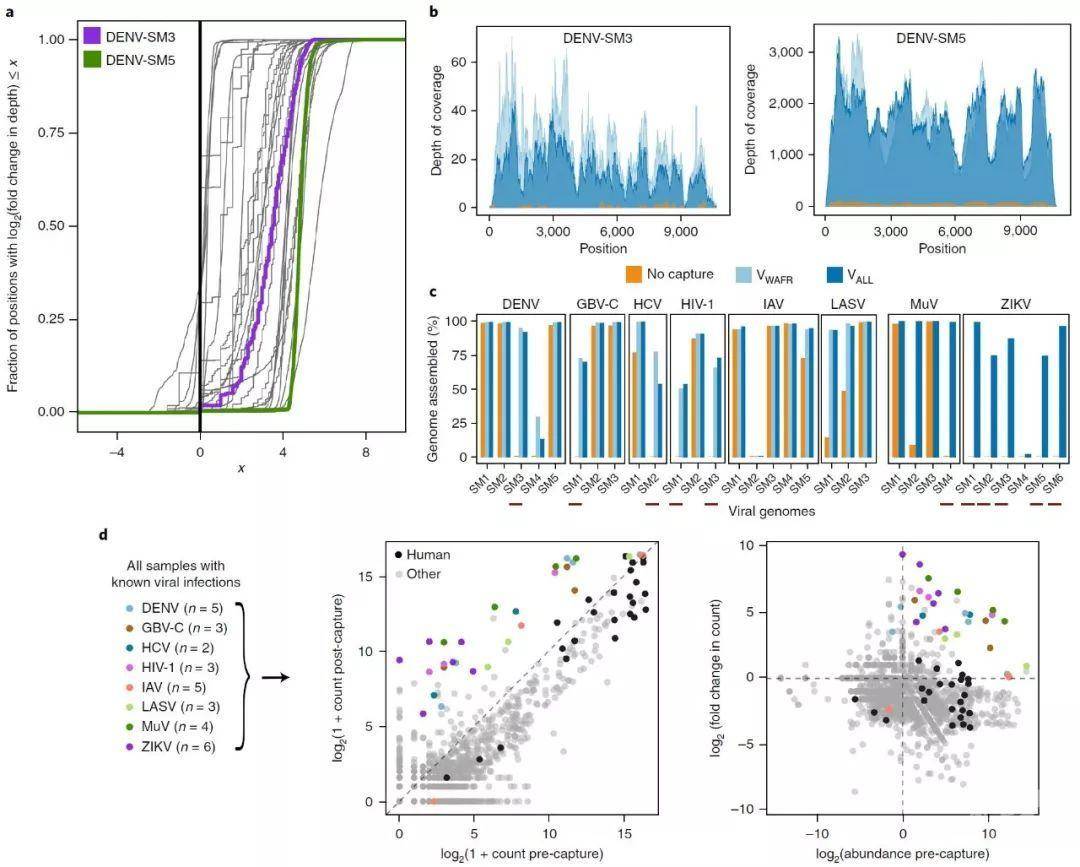
a,通过对30例患者和已知病毒感染的环境样本进行VALL捕获,病毒基因组的富集的测序深度分布。每条曲线代表在此测序的31个病毒基因组中的一个(一个样本包含两种已知病毒)。在基因组的每个位置,捕获后测序深度除以捕获前深度,绘制的曲线是这些倍数变化值的对数的经验累积分布。一条完全上升到黑色垂直线右侧的曲线说明了整个基因组的富集;曲线越垂直,富集越均匀。病毒基因组DENV-SM3(紫色)和DENV-SM5(绿色)的测序深度在b中显示得更详细。
b, 在两个样本中测序整个DENV基因组的深分布度。DENV-SM3(左图)在捕获前的信息性序列很少,不产生基因组组装,但在捕获后可行。DENV-SM5(右)的确在捕获前产生了一个基因组组装,捕获后深度增加。
c, 在30个样本中,每个病毒基因组的百分比都清晰地聚集在一起,其中有8个已知的病毒感染。捕获前(橙色)、捕获后用VWAFR(浅蓝色)和捕获后用VALL(深蓝色)显示。样本下面的红色条表明,我们在捕获前无法装配任何重叠群,但在捕获后我们能够装配至少部分基因组(>50%)。
d,左边,在用VALL捕获之前和之后30个已知病毒感染的样本中检测到的每个物种的序列次数。将每个样本中的读数减至200,000个序列。每个点代表一个样本中检测到的一个物种。对于每个样本,先前通过另一种分析方法检测到的病毒都被着色。人类样本中的人源序列匹配显示为黑色。对在捕获前检测到的每一种物种的丰度,以及用VALL捕获后,这些样本进行的富集倍数变化。通过将每个物种的捕获前序列计数除以水对照总合序列来计算丰度。人类和病毒物种的着色如左图所示。
Fig. 2 | Improvement in genome coverage and assembly, and shift in metagenomic distribution after capture.
a, Distribution of the enrichment in read depth, across viral genomes, provided by capture with VALL on 30 patient and environmental samples with known viral infections. Each curve represents one of the 31 viral genomes sequenced here (one sample contained two known viruses). At each position across a genome, the post-capture read depth is divided by the pre-capture depth, and the plotted curve is the empirical cumulative distribution of the log of these fold-change values. A curve that rises fully to the right of the black vertical line illustrates enrichment throughout the entirety of a genome; the more vertical a curve, the more uniform the enrichment. Read depth across viral genomes DENV-SM3 (purple) and DENV-SM5 (green) is shown in more detail in b.
b, Read depth throughout the DENV genome in two samples. DENV-SM3 (left) has few informative reads before capture and does not produce a genome assembly, but does following capture. DENV-SM5 (right) does yield a genome assembly before capture, and depth increases following capture.
c, Percent of each viral genome unambiguously assembled in the 30 samples, which had eight known viral infections across them. Shown before capture (orange), after capture with VWAFR (light blue), and after capture with VALL (dark blue). Red bars below samples indicate ones in which we could not assemble any contig before capture but in which, following capture, we were able to assemble at least a partial genome (> 50%).
d, Left, number of reads detected for each species across the 30 samples with known viral infections, before and after capture with VALL. Reads in each sample were downsampled to 200,000 reads. Each point represents one species detected in one sample. For each sample, the virus previously detected in the sample by another assay is colored. Homo sapiens matches in samples from humans are shown in black. Right, abundance of each detected species before capture and fold change upon capture with VALL for these samples. Abundance was calculated by dividing pre-capture read counts for each species by counts in pooled water controls. Coloring of human and viral species is as in the left panel.
图3. 改进样品多样性的检测和保留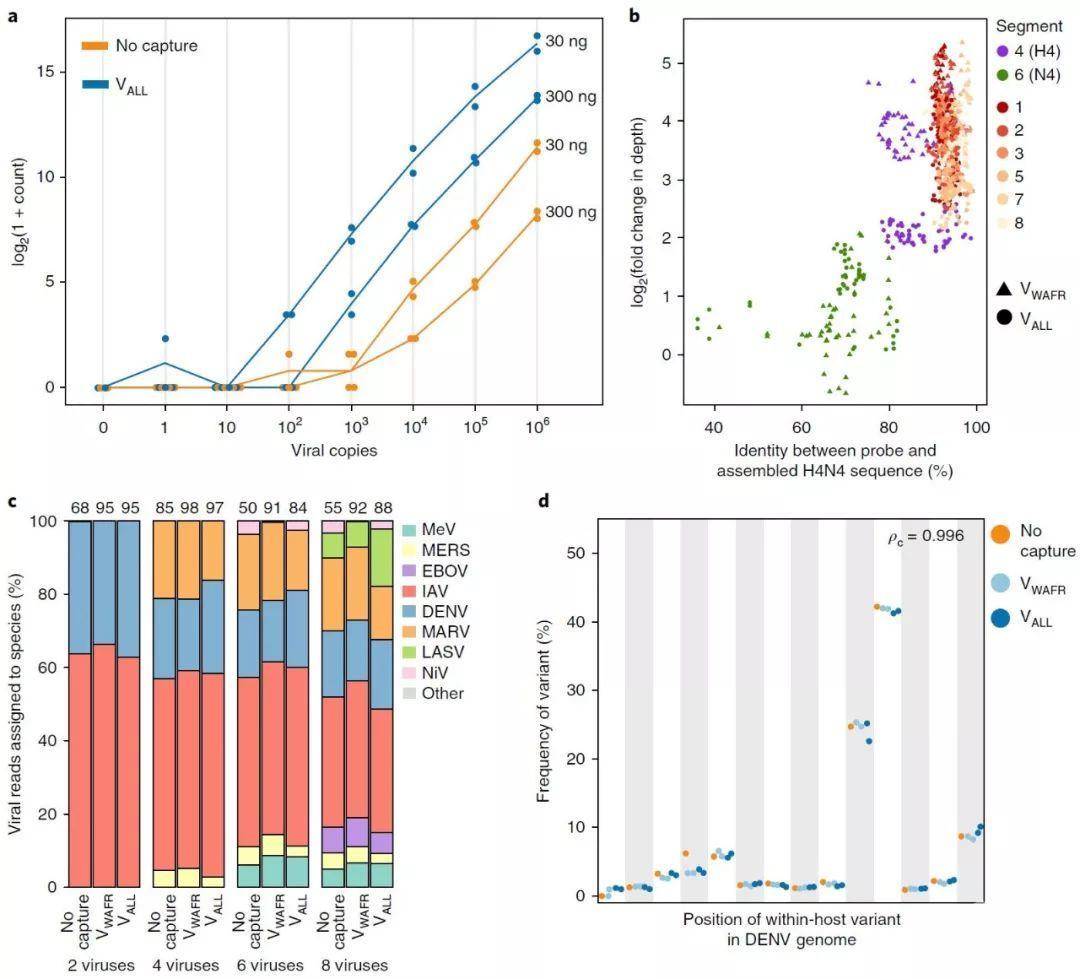
A,在两种人类RNA背景下,病毒输入稀释序列中测序的病毒材料量。对于每种输入拷贝的选择、背景数量和捕获方法,都有n=2个技术复制(n=1为负对照无重复)。每一个点表示从一个重复中测序出的20万个唯一病毒读取的数量;该行通过重复的平均值。每行右侧的标签指示背景材料的起始量。
b,探测-目标相似性和测序深度与富集程度之间的关系,如IAV样品用VALL和VWAFR捕获后在H4N4 (IAV-SM5)上结果。每个点代表IAV基因组中的一个窗口。探针和组装的H4N4序列之间的一致性是该窗口中的序列与其对应的探针序列的前25%之间的一致性度量(有关详细信息,请参见方法)。富集倍数的变化在窗口上取平均值。在VALL和VWAFR的设计中没有包括N4亚型的第6段(N)的序列。
c, 捕获对样本内共感染估计频率的影响。将2种、4种、6种和8种病毒的RNA加入健康人血浆中提取的RNA中,然后用VALL和VWAFR捕获。上面的值是所有病毒性序列数量的百分比。MEV为麻疹病毒,MES为中东呼吸综合征冠状病毒,MARV为马尔堡病毒,NIV为尼帕病毒。我们没有使用VWAFR探针集检测到Niv,因为该设计中不存在这种病毒。
d,捕获对宿主内变异估计频率的影响,显示在三个DENV样本的位置:DENV-SM1、DENV-SM2和DENV-SM5。在同一个库的n=2个拷贝上使用VALL和VWAFR进行捕获。ρc表示捕获前后频率的一致性相关系数。
Fig. 3 | Characterizing improvement in detection and preservation of within-sample diversity.
a, Amount of viral material sequenced in a dilution series of viral input in two amounts of human RNA background. There are n = 2 technical replicates for each choice of input copies, background amount, and use of capture (n = 1 replicate for the negative control with 0 copies). Each dot indicates the number of unique viral reads, among 200,000 in total, sequenced from a replicate; the line is through the mean of the replicates. The label to the right of each line indicates the amount of background material.
b, Relationship between probe–target identity and enrichment in read depth, as seen after capture with VALL and with VWAFR on an IAV sample of subtype H4N4 (IAV-SM5). Each point represents a window in the IAV genome. Identity between the probe and assembled H4N4 sequence is a measure of identity between the sequence in that window and the top 25% of probe sequences that map to it (see Methods for details). Fold change in depth is averaged over the window. No sequences of segment 6 (N) of the N4 subtypes were included in the design of VALL or VWAFR.
c, Effect of capture on the estimated frequency of within-sample co-infections. RNA of 2, 4, 6, and 8 viral species was spiked into RNA extracted from healthy human plasma and then captured with VALL and with VWAFR. Values on top are the percent of all sequenced reads that are viral. MeV is measles virus, MERS is Middle East respiratory syndrome coronavirus, MARV is Marburg virus, and NiV is Nipah virus. We did not detect NiV using the VWAFR probe set because this virus was not present in that design.
d, Effect of capture on the estimated frequency of within-host variants, shown in positions across three DENV samples: DENV-SM1, DENV-SM2, and DENV-SM5. Capture with VALL and VWAFR was performed on n = 2 replicates of the same library. ρC indicates the concordance correlation coefficient between the pre- and post-capture frequencies.
图4. 利用捕获技术的基因组应用:2018年拉沙热爆发未分类样本中感染的测序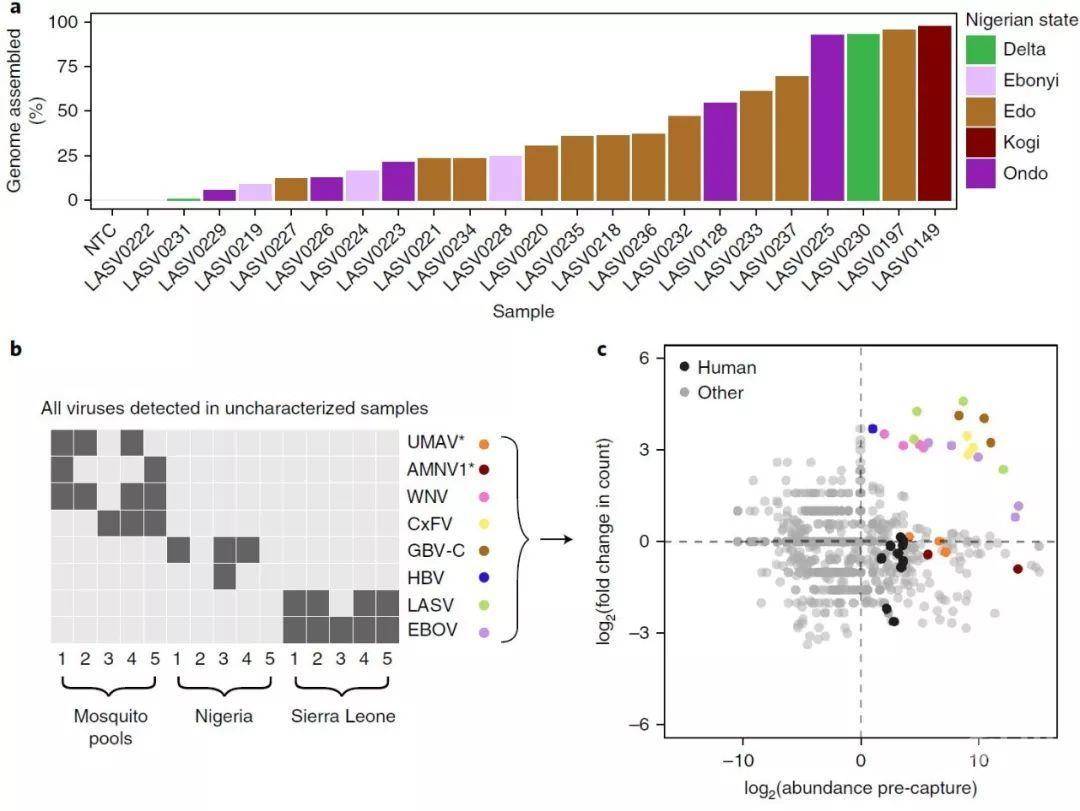
a. 2018年拉沙热爆发的23个样本中,在使用VALL后收集了拉沙病毒基因组组装的百分比。在装配前,读数被降低到200,000次。按照样品来自尼日利亚的来源着色,柱状图展示组装的比例。
b,在用VALL捕获后,存在于未知的蚊子群体和来自尼日利亚和塞拉利昂的人血浆样本中的病毒种类。物种上的星号表示那些不VALL所捕获的。检测到的病毒包括尤马蒂病毒(UMAV)、Alphamesonivirus 1 (AMNV1))、西尼罗河病毒(WNV)、库蚊黄病毒(CxFV)、GBV-C、乙型肝炎病毒(HBV)、LASV和EBOV。
c, 捕获前检测到的所有物种的丰度,以及在未知样品池中用VALL捕获后的倍数变化。如图2d所示计算丰度。每个样本中存在的病毒种类(见b)都被着色,人类血浆样本中的人源匹配物以黑色显示。
Fig. 4 | Genomic applications using capture: sequencing from the 2018 Lassa fever outbreak and of infections in uncharacterized samples.
a, Percent of the LASV genome assembled, after use of VALL, among 23 samples from the 2018 Lassa fever outbreak. Reads were downsampled to 200,000 reads before assembly. Bars are ordered by amount assembled and colored by the state in Nigeria that the sample is from.
b, Viral species present in uncharacterized mosquito pools and pooled human plasma samples from Nigeria and Sierra Leone after capture with VALL. Asterisks on species indicate ones that are not targeted by VALL. Detected viruses include Umatilla virus (UMAV), Alphamesonivirus 1 (AMNV1), West Nile virus (WNV), Culex flavivirus (CxFV), GBV-C, hepatitis B virus (HBV), LASV, and EBOV.
c, Abundance of all detected species before capture and fold change upon capture with VALL in the uncharacterized sample pools. Abundance was calculated as described in Fig. 2d. Viral species present in each sample (see b) are colored, and H. sapiens matches in the human plasma samples are shown in black.
来源:meta-genome 宏基因组
原文链接:https://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247493187&idx=4&sn=ead41d4c2fa295e08b723e6bd46875c3&chksm=faba0cf2cdcd85e485cfa627fb092f72f0189f66143cf4ceab4a277d67dd383e4909e6211616#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
MetaQuast:评估宏基因组拼接
Prokka:快速原核基因组、宏基因组基因注释

Nature子刊:Salmon不比对快速宏基因组基因定量

真菌基因组和宏基因组学讲习班

PICRUSt2预测宏基因组功能

Nature子刊:Salmon不比对快速宏基因组基因定量
metaProdigal:宏基因组序列中的基因和翻译起始位点预测

宏基因组如何指导微生物分离培养
MetaQuast:评估宏基因组拼接
宏基因组理论教程2扩增子分析


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号