来源:X一MOL资讯
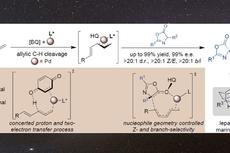
氟是药物化学家最喜欢的原子之一,尽管体积与氢差不多,但其生物效应却可能对候选药物的物理化学性质、代谢稳定性、透膜性等产生神奇的变化。然而,与C-H键不同(可通过均裂或异裂进行活化)的是,C-F键由于键能太高几乎不能被均裂。同样的,由于键能高、氟离子的碱性强,通常需要强Lewis酸才能发生C-F键异裂,这也与药物化学家利用氟原子屏蔽易代谢位点是一致的。另一方面,对C(sp
3)-H键进行对映选择性官能团化已有相当成熟的方法,比如羰基α-位不对称锂化、苄位不对称C-H键活化、卡宾插入或氮宾插入以及导向的C-H键活化等等(下图a)。然而,对偕二氟亚甲基单元进行对映选择性的取代单氟原子尚未见报道,这是因为这样的C-F键具有更高的键能。
面对这座看似不可逾越的“大山”,近日美国加州大学伯克利分校的John F. Hartwig教授课题组做出了新的突破,给后来人提供了一种解决思路。他们设想:通过低价过渡金属催化剂活化二氟亚甲基的相邻官能团(如3-取代的3,3-二氟丙烯上的双键)以活化二氟亚甲基单元(推效应);然后通过亲氟的阳离子接受其中一个氟离子并产生单氟π-烯丙基中间体(拉效应);最后亲核试剂进攻该中间体,便能得到叔烷基氟化物(下图c),该产物的对映选择性来自于π-烯丙基-催化剂复合物的立体环境。虽然早在2018年该课题组就发现,手性铱配合物能够催化氟代烯丙基的对映选择性取代,但该反应是通过转化限制的氧化加成(turnover-limiting oxidative additions)或放热/可逆的氧化加成(endothermic and reversible oxidative additions)实现的,并且氟代烯丙基化合物也需要三氟乙酸酯或者磷酸酯作为离去基团(Angew. Chem. Int. Ed., 2018, 57, 13125-13129)。在最近的工作中,他们发现环金属化的铱催化剂和叔丁醇锂或硅醚化合物能够通过上述“推-拉协同效应(push-pull cooperation)”以高收率、高区域选择性和化学选择性实现烯丙基偕二氟化合物的高对映选择性去单氟官能团化,相关结果发表在Nature 上。
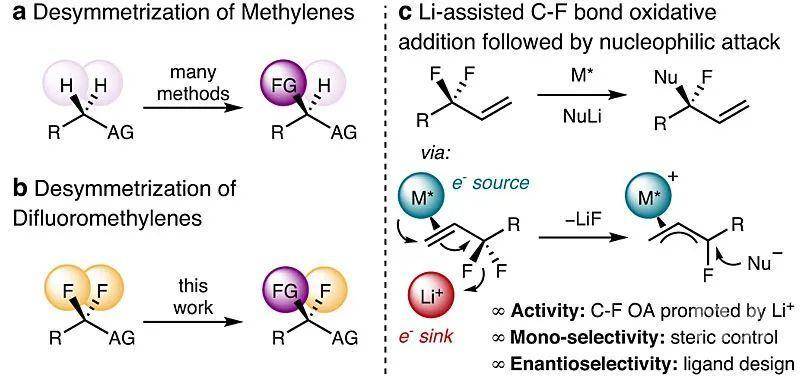
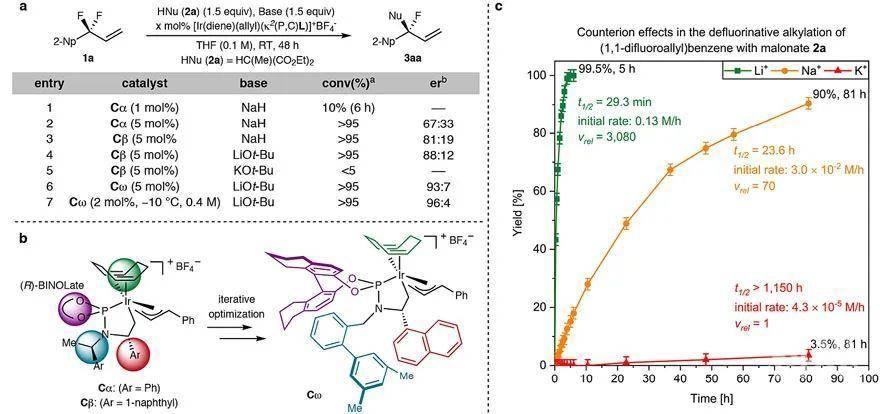
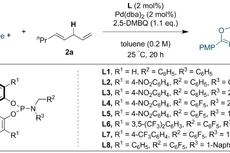
亚甲基去对称化策略。图片来源:Nature首先,研究人员以2-(1,1-二氟烯丙基)萘(1a)为底物进行条件优化。在铱催化剂Cα(1 mol%)作用下与甲基丙二酸二乙酯的钠盐(从NaH制备)反应6 h后,能以> 99:1的er值转化为目标产物3aa,但收率仅10%,延长反应时间并增加催化剂用量能提高收率,但对映选择性明显降低(er 值:67.1:32.9,下表entries 1和2),而改用催化剂Cβ能稍微提高er值(81:19,下表entry 3)。这使得作者考虑是否“拉”的效应还不够强,因此进一步筛选亲氟活化剂。值得注意的是,当用丙二酸锂进行反应时,反应速率要比钠盐快44倍(对映选择性(88:12 er vs 82:18 er)也稍好,下表entry 4),比钾盐快3080倍(下图c),这表明抗衡离子有助于C-F键的氧化加成。通过改变催化剂的结构和反应条件,最终获得最优条件——即2 mol%催化剂Cω、叔丁醇锂和-10 ℃——er值提高到96:4(下表entry 7)。
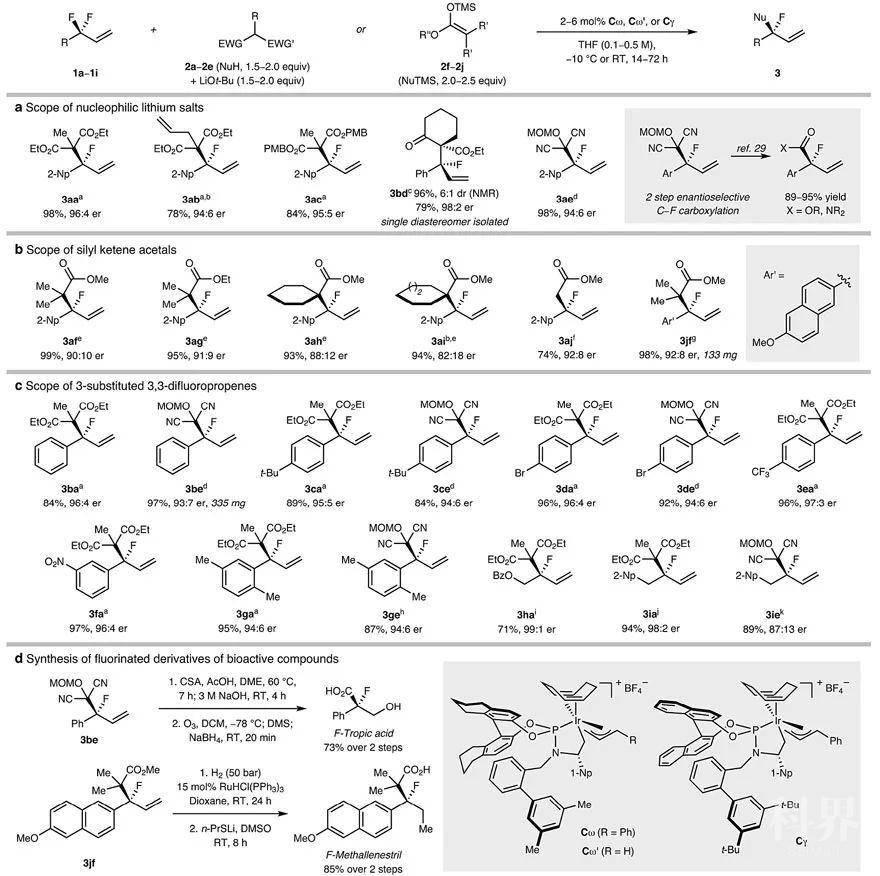
条件优化。图片来源:Nature在最优反应条件下,研究人员进行了大量的底物拓展。首先,除了丙二酸酯(1a-1c)外,丙二腈1e也能很好地进行相应的转化(LiBr作为添加剂),以较高的收率和优异的对映选择性得到目标产物(3aa-3ac、3ae),并且丙二腈可作为酰基负离子等价物,可以在酸性条件下进一步转化为酯或酰胺,例如通过两步法的策略便可实现3-取代的3,3-二氟丙烯中一个C-F键的对映选择性羧化(下图a)。有意思的是,前手性β-酮酸酯也能兼容该反应,以较好的非对映选择性(dr值:6:1)和优异的对映选择性(er值:98:2)得到具有两个连续且完全取代的立体中心——叔烷基氟化物3bd,并以79%的收率分离出3bd的主要非对映异构体。其次,硅基也能作为亲氟活化剂,环状、非环状以及未取代的烯醇硅醚(2f-2j)都可以在铱催化剂下直接与1a或1j反应,此时使用Cγ产生的对映选择性要比Cω高。第三,不同电性的亲电试剂(3a-3i)对反应的产率和对映选择性影响不大。对于烷基取代的3,3-二氟丙烯类底物,反应需要升温(3ha)或添加三氟甲磺酸钡作为Lewis酸活化剂(3ia、3ie)。最后,研究人员用该方法制备了两个生物活性物质的衍生物,如F-莨菪酸和F-美沙雌酸,证实了该方法不仅底物范围广,而且在药物发现中具有潜在的应用价值。
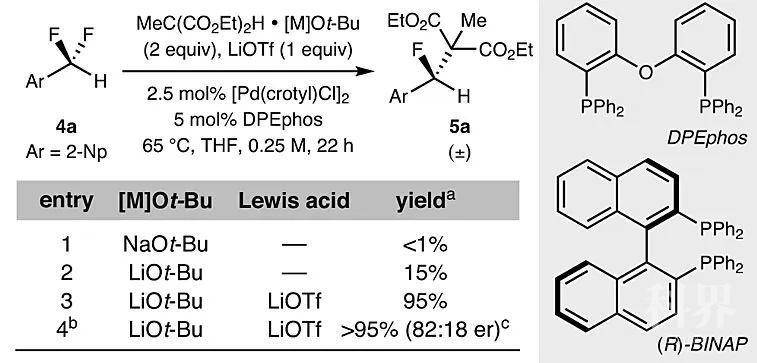
底物扩展和应用。图片来源:Nature由此可见,采用这种“推-拉”活化的方式能够对烯丙位二氟亚甲基进行选择性的C-F键活化,那么对活性较低的苄位C-F键效果如何呢?可喜的是,2-(二氟甲基)萘在[Pd(crotyl)Cl]
2、DPEphos和LiOTf的作用下与甲基丙二酸二乙酯•LiOt-Bu复合物反应,能以> 95%的收率和82:18的er值转化为单氟化合物5a。
选择性苄位C-F键活化。图片来源:Nature要想探究反应机理,必须要搞清楚速率控制步骤、对映选择性控制步骤以及绝对构象的起源。首先,研究人员分析了催化剂和底物组成的静息状态(resting state)的
19F和
31P NMR,结果显示为80:20的非对映体混合物,并且互变非常快(t
1/2forward= 0.88s和t
1/2reverse= 0.23s)。静息状态经C-F键的氧化加成生成π-烯丙基中间体,后者与丙二酸酯锂盐的反应也是非常快(t
1/2< 20 sec),在2 min内就能达到> 98%的转化率,因此这两个过程都不是决速步。另外,催化剂Cω与丙二酸酯锂盐反应迅速形成Ir(I)物种,即使延长反应时间,也不与副产物氟化锂发生反应。这些数据暗示C-F键的氧化加成可能是决速步。
其次,产物的绝对构象是如何产生的呢?这显然与烯烃配位到催化剂的过程无关,因为这个resting state是快速互变的。那与亲核进攻有关吗?研究人员认为这种情况必须涉及可逆的氧化加成或π-σ-π互变。但他们发现3-氟烯丙酯和3-取代的-3,3-二氟丙烯在相同的条件下却得到互为对映体的产物(下图c),这表明π-烯丙基中间体不可能发生π-σ-π互变,这也被能量计算所证实(翻转能垒高达21.9 kcal/mol,下图d)。因此,氧化加成是决定对映选择性的关键步骤。

机理研究。图片来源:Nature至于绝对构型的起源,研究人员结合密度泛函理论计算和四象限模型进行解释。如上图e所示,催化剂的环庚二烯(COD)处于东北象限(NE),磷酸基团处于西北象限(NW),含N基团位于西南象限(SW),3-氟烯丙酯和3-取代的-3,3-二氟丙烯的双键从面上接近催化剂时,最大的基团(分别是Ph和PhCF
2)需要放置在位阻最小的东南象限(SE),才是能量上最有利的。也就是说,3-氟烯丙酯和3-取代的-3,3-二氟丙烯以不同的方式与催化剂结合,从而导致产生不同构型的产物。
基于以上研究,研究人员提出了可能的反应机理(上图a):首先,阳离子Ir(III)烯丙基预催化剂Cω与丙二酸锂快速反应,释放出Nu-allyl和LiBF
4,并与1 equiv的起始原料(3-取代的3,3-二氟丙烯)结合,生成非对映异构的Ir(I)烯烃络合物的混合物。随后通过抗衡离子辅助的氧化加成生成exo(major)或endo(minor)Ir(III)-π-烯丙基络合物,后者被丙二酸酯迅速捕获生成Ir(I)产物络合物。最后,用等量的起始原料进行配体解离得到取代产物。
总结
Hartwig课题组通过低价过渡金属和亲氟阳离子组合产生的“推-拉效应”使原本惰性的二氟亚甲基能够对映选择性地去对称化,这项研究为化学工作者制备手性叔烷基氟化物提供了新的思路,由于氟的特殊性,或许还能为药物发现和改进提供一条高效、简便的方法。
来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657641335&idx=1&sn=8499fa02a45966a32a1c5d7684fbf752&chksm=80f84aa7b78fc3b1996455a484d630a51155764bc0343cec23835f971a9b102d32c74d2c2899&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn



 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号