科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-07-12
来源:生物医药工程智库
2020年6月,《中华人民共和国药典》(简称《中国药典》)2020年版正式上市,2020版三部《中国药典》首次新增了9402《生物制品稳定性试验指导原则》,这是一份第十一届药典委送给生物制品领域的药界人士的惊喜大礼。
生物制品相对比较特殊,一般情况下,很难直接套用化学药、中药类的指导原则进行实际操作,本文从为何要开展稳定性研究?如何开展稳定性研究?等方面探讨生物制品稳定性研究主要内容、存在问题及应对措施。
一、为何要开展稳定性研究?
稳定性研究是直接决定一种生物制品是否能够成为上市产品的关键性研究,稳定性研究是贯穿于整个药品研发阶段和支持药品上市及上市后研究的重要内容,是产品有效期设定的依据,是评价药品有效性和安全性的重要指标之一,可以用于对产品生产工艺、制剂处方、包装材料选择合理性的判断,同时也是产品质量标准制订的基础。
对于生物制品药品而言,其活性成分蛋白质的分子构型和生物活性的保持都依赖于与其本身所处的微环境,如温度、光照、离子浓度和机械剪切力等存在着密切的关系。
因此生物制品药物应开展严格全面的稳定性研究,目前国内有部分处于新药研发阶段的生物制品企业对于稳定性研究还是重视不够,申报资料关于稳定性研究比较简单,就是汇总一下影响因素试验、加速试验和长期试验的数据,然后简单做一个趋势图,不能给药品有效期的确定提供充分的支持,除在产品研发阶段需要进行稳定性研究,已上市生物制品在稳定性研究常见的缺陷如未在稳定性考察方案在对重大偏差、变更等情况列入稳定性考察内容,生物医药企业以下变更情况建议开展稳定性研究,如:处方变更、西林瓶或胶塞等内包装容器变更、剂型变更、生产变更(包括工艺变更,设备变更、场所变更等)、生产规模变更等;影响安全性或有效性的偏差等情况。
二、如何开展稳定性研究?
稳定性考察应当制定考察方案,本文梳理了2020版三部《中国药典》和2010版GMP通则要求,概述如下:
2010版GMP通则考察方案应当至少包括以下内容:考察批次数;检验方法及依据;检验项目及合格标准;容器密封系统的描述;试验间隔时间;贮存条件;
2020版三部《中国药典》9402《生物制品稳定性试验指导原则》:稳定性试验应制订产品稳定性评价的详细方案。该方案能支持产品建议的贮存条件和有效期。方案应包含证明产品稳定性的试验类别、试验样品、试验项目、试验条件、试验时间和结果分析等内容。
稳定性研究另一常见缺陷是稳定性方案设计不周,2020版三部《中国药典》和2010版GMP通则都要求稳定性考察应当有考察方案,但要求内容不完全一致,如何涉及稳定性方案才能更合规呢?
9402《生物制品稳定性试验指导原则》规定的稳定性方案内容更具有可操作性,包括要素更能体现稳定性考察的关键要素,2010版GMP通则重点强调的是稳定性考察项目及其检验方法,建议制定生物制品稳定性考察方案时至少包括试验类别、试验样品(考察批次)、试验项目(及合格标准)、试验条件(含容器密封系统的描述)、试验时间和结果分析等内容,不能有漏项。
三、生物制品稳定性研究有哪些类别?
按照中国药典2020版要求,我国的生物制品稳定性研究可以分为影响因素试验、加速试验和长期试验,不同的试验类别考察条件、目的和用途是不一样的,主要差别如下表所示:

四、生物制品哪些考察对象需要做稳定性研究?
生物制品稳定性研究考察对象(或称试验样品)主要包括原液、成品及产品自带的稀释液或重悬液,对因不能连续操作而需保存一定时间的中间产品也应进行相应的稳定性研究。
本次药典发布的指导原则与原CFDA2015年4月15日发布实施的《生物制品稳定性研究技术指导原则(试行)》考察对象略有不同,该版本要求对因不能连续操作而需保存一定时间的中间产物,2020版三部《中国药典》9402《生物制品稳定性试验指导原则》正文其它部分都是使用中间产物的术语,那么是在考察对象故意修订为中间产品还是需要勘误,这个待观察药典委的后续说明;
中间产品和中间产物专业术语是有差距,笔者建议生物制品使用中间产物较合适,2010版GMP通则中间产品是指完成部分加工步骤的产品,尚需进一步加工方可成为待包装产品;而原CFDA2015年4月15日发布实施的《生物制品稳定性研究技术指导原则(试行)》中间产物是指生产过程中形成的、为下一步工艺所用的物质,不包括原液。
生物制品纯化层析过程中有一些中间产物因不能连续操作而需保存一定时间,需要按照指导原则进行相应的稳定性研究。
五、生物制品稳定性研究有哪些重点考察项目?
生物制品稳定性评价指标较为复杂,应根据不同品种的成分特性开展稳定性实验工作,应尽量使用适当的理化、免疫化学方法对生物制品的活性成分进行定量检测,降解产物的分析也是稳定性试验的重要组成部分,检验项目,如检验项目少于成品质量标准所包含的项目,应当说明理由,重点考察项目原则需要考虑以下几个方面:
(1) 检测项目应包括产品敏感的,且有可能反映产品质量、安全性和/或有效性的考查项目,如生物学活性、纯度和含量等。其中生物学活性/效价生物学活性/效价是生物制品稳定性试验中的关键评价指标;生物制品的纯度应采用多种原理的分析方法进行综合评估。
(2) 根据产品剂型的特点,应考虑设定相关的考察项目,如外观、可见异物、不溶性微粒、pH值、注射用无菌粉末的水分、无菌检查等;
(3) 气品添加剂(如稳定剂、抑菌剂)或赋形剂在制剂的有效期内也可能降解,若有迹象表明这些物质的降解对药品质量有不良影响时,应在稳定性试验中加以监测。
(4) 稳定性试验中还应考虑到包装容器和密闭系统可能对样品具有潜在的不良影响,在试验设计过程中应关注此方面。
(5) 为了保证稳定性研究结果的真实可靠,稳定性试验的检验方法应经过方法学的验证,包括但不限于专属性、准确性、精密度、灵敏度等。
六、生物制品稳定性研究考察条件如何考虑?
生物制品稳定性研究应根据研究目的和产品自身特性对研究条件进行摸索和优化,稳定性研究条件应充分考虑到今后的贮存、运输及其使用的整个过程,并考虑根据对各种影响因素(如温度、湿度、光照、反复冻融、振动、氧化、酸碱等相关条件)的初步研究结果,制定长期、加速和强制条件试验等稳定性研究方案,重点考察条件需要考虑以下几个方面:
1. 温度
(1) 影响因素试验:温度应达到可以观察到样品发生降解并超出质量标准的目的;
(2) 加速试验:温度条件一般介于长期与强制条件试验之间,通常可以反映产品可能短期偏离于实际保存条件的情况。
(3) 长期试验:温度条件应与实际保存条件相一致。
(4) 拟常温贮存的产品,长期试验建议采用的温度为25℃±2℃或30℃±2℃;加速试验建议采用的温度为40℃±2℃;
(5) 拟冷藏贮存的产品,长期试验建议采用的温度为5℃±3℃;加速试验建议采用的温度为25℃±2℃;
(6) 拟冷冻贮存的产品,长期试验建议采用的温度为-20℃±5℃;加速试验建议采用的温度为5℃±3℃或25℃±2℃;
(7) 需更存储在-20℃以下的产品,可根据其特点制定合适的试验温度。
2. 湿度
如能证明包装容器与密闭系统具有良好的密封性能,则不同湿度条件下的稳定性试验可以省略;否则,需要开展相关研究。
3. 包装容器与密闭系统
生物制品可能会与密闭系统相互作用而发生变化。通常应考虑液体制剂与密闭系统的相互作用,应将样品以倒立放置或水平放置、正立放置两种情况进行稳定性试验,以确定密闭系统对产品的影响,原则上液体制剂与密闭系统应充分接触,不同密闭系统的产品应分别进行稳定性试验。
如果产品为多次使用的包装,应模拟实际使用情况对样品进行稳定性试验,确保多次使用后产品的稳定性仍符合标准。
4. 反复冻融
对于需冷冻保存的原液、中间产物,应验证其在多次反复冻融条件下产品质量的变化情况。
5. 运输条件
生物制品通常要求冷链保存和运输,应对产品的运输条件进行相应的模拟试验。稳定性试验时,应充分地考虑运输路线、交通工具、运输距离、运输时间、装载模式、外界环境以及运输时可能遇到的最差条件。
通过试验,应确认产品在运输过程中处于拟定的保存条件下可以保持产品的稳定性,并评估产品在短暂地脱离拟定保存条件下对产品质量的影响。对于需要冷链运输的产品,应尽可能对产品脱离冷链的温度、次数、总时间等制定相应的要求。
6. 其他
对于需要复溶、稀释的产品,应根据具体情况对使用过程中涉及的条件设计相应的稳定性试验,如某些情况下,根据产品特点设计对于光照、振动和氧化等条件的试验。
另外,液体制剂在稳定性研究中还应考虑到产品的放置方向,如正立、倒立或水平放置等。模拟实际使用情况的研究应考虑产品使用、存放的方式和条件,如注射器多次插入与抽出的影响等。
对于一些生物制品,如用于多次使用的、单次给药时间较长的(如静脉滴注)、使用前需要配制的、特殊环境中使用的(如高原低压、海洋高盐雾等环境)、以及存在配制或稀释过程的小容量剂型等特殊使用情况的产品,应开展相应的稳定性研究,以评估实际使用情况下产品的稳定性。
七、生物制品稳定性研究研究时间点如何考虑?
稳定性研究中考察时间的长短及放置条件应当充分考虑到今后的贮藏、运输及其使用的整个过程,在某些特殊情况下,可灵活调整检测时间,比如,基于初步稳定性研究结果,可有针对性的对产品变化剧烈的时间段进行更密集的检测,产品有效期的制定应根据长期稳定性研究结果设定;
(1) 如果产品的预定有效期在1年或1年以内,长期稳定性研究时间点原则上应在前3个月每月试验1次,以后每3个月试验1次,即0、1、2、3、6、9、12月。
(2) 如果产品的预定有效期在1年以上,长期稳定性研究时间点原则上应在第1年每3个月试验1次,第2年每6个月试验1次,以后每年试验1次,即0、3、6、9、12、18、24、36月,如效期长于,36个月,则每年一次直至有效期止。
(3) 影响因素和加速稳定性研究应观察到产品不合格。
(4) 长期稳定性研究应尽可能做到产品不合格为止。
来源:BOPUZHIKU1314 生物医药工程智库
原文链接:https://mp.weixin.qq.com/s?__biz=MzU5Mjc3ODgzMQ==&mid=2247493165&idx=1&sn=f6bb7f9255d3edd95ea89d1f64678dd4&chksm=fe18250dc96fac1bc6bd65417ff42b07448389a021cc00c4dae478011262e5687f13eeb40e66#rd
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

提升生态系统质量和稳定性
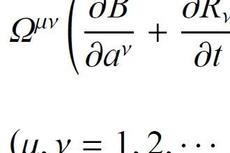
陈菊等:Birkhoff系统稳定性的动力学控制

院士风采|薛禹胜:稳定性理论及电力系统自动化专家

生物同质化可削弱生态系统的稳定性
AI新应用——预测行星系统的稳定性
2016世界生命科学大会在京举行
中国毒理学会2017年活动计划
第七届全国微生物遗传学学术研讨会圆满结束

【优文报告】中低压柔性直流配电系统稳定性分析模型与机理研究综述

第十五届全国植物基因组学大会在安徽顺利召开


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号