科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-07-24
来源:Nextomics
2020年7月,PB和ONT技术结合在人类基因组上取得突破性进展。国际端粒到端粒联盟(Telomere-to-Telomere Consortium,T2T)于Nature杂志发表了重磅文章“Telomere-to-telomere assembly of a complete human X chromosome”,利用Nanopore ultra-long、PacBio、10X Genomics等多种测序技术,成功组装出首个端粒到端粒(Telomere-to-Telomere)的无缺口人类X染色体完成图[1]。此项工作引发了基因组学领域内学者对Telomere-to-Telomere(T2T)基因组完成图的热烈讨论。斯坦福大学Tim教授在推特表示“在人类基因组草图公布20年后,我们有了第一个完整组装的人类染色体”,随后,国际语音识别和人工智能专家Daniel Povey留言表达了对这项工作的认可和期待。
从2001年发布首个人类参考基因组至今,已陆陆续续发表了一系列参考基因组版本。事实上,人类基因组尚未解决的区域还有很多,比如着丝粒周围Mb级别的微卫星区域,近端着丝粒短臂上的rDNA区域,长达几百Kb的片段重复等。因此,在人类及其他动植物基因组领域,实现基因组完成图是一个终极目标。而在基因组不断完善的过程中,测序技术的发展起着举足轻重的作用。一代测序单碱基准确性高达99.999%,读长可达1kb,但是通量太低,项目周期很长;二代测序(Next-Generation Sequencing)也称高通量测序技术,最大的特点是通量很高成本低,但是二代短读长极大地限制了其在基因组重复序列区域和同源基因辨别方面的发挥。PacBio(PB)和 Oxford Nanopore Technologies (ONT)公司开发的三代测序技术迎难而上,近几年,科学家们基于三代长读长的特性,将人类基因组组装的连续性提升到一个新水平。
ultra-long reads对于组装着丝粒和端粒附近的大量的串联重复序列具有重要的参考价值,为最终获得高精度和完整的基因组序列奠定了良好的基础。
2018年,英国伯明翰大学微生物学与感染研究所Nicholas J Loman团队联合英国诺丁汉大学生命科学学院Matthew Loose团队,在Nature Biotechnology发表了题为“Nanopore sequencing and assembly of a human genome with ultra-long reads”的研究成果,研究者用MinION对人类GM12878 Utah/Ceph细胞系进行测序和组装,理论深度为30X,De novo组装得到NG50约3M。同时,研究者提出一套方法通过ONT技术获得ultra-long reads(N50>100kb,最大达882kb),把这部分5X的超长读长数据加进来之后,使得组装的连续性增加了一倍多(NG50 约6.4 Mb)。最终组装得到的基因组大小为2.867G,覆盖了参考基因组的85.8%。并且,实现了4 Mb主要组织相容性复合体(MHC)位点完整组装和分相,测量端粒重复长度,并填补了参考基因组GRCh38中的Gap[2]。
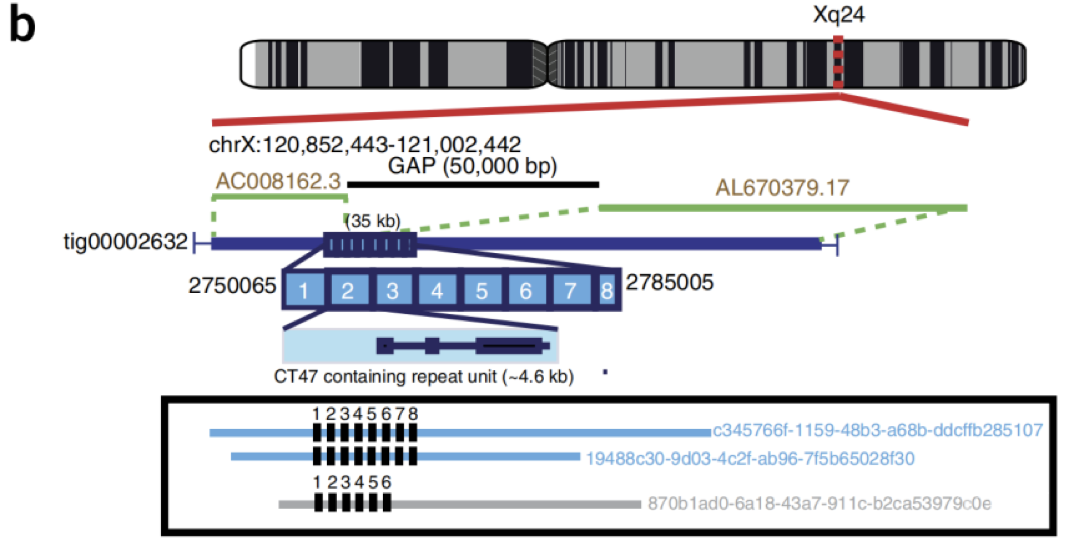
图1 超长读长数据填补参考基因组的Gap
HiFi reads结合HiCanu完成准确度超99.999%,contig N50达到77M的人类基因组组装结果。
2020年,来自美国NIH人类基因组研究所的研究团队预印发表了他们题为“HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads”最新成果,文章中使用30X PacBio HiFi Reads和HiCanu组装工具(专门针对PacBio HiFi Reads优化了组装流程),对人CHM13细胞系的基因组进行了组装,单碱基准确性超过99.999%(Q50),contig N50达到77M。研究中还重点探索了HiCanu结合HiFi 数据的方式,在基因组单倍型多样性、主要组织相容性复合体(MHC)变异、复杂片段重复序列、染色体上卫星区域、着丝粒组装等方面的效果提升。HiFi数据在解决大型高度相似(但不相同)的重复方面表现出色,但对于像X染色体这样,存在大片段重复(>20kb),重复序列几乎相同(99.9%)的区域,HiFi数据因为不够长没办法充分分辨[3]。因此仅凭PB HiFi reads想要实现端粒到端粒的完整组装,还有一定的难度。
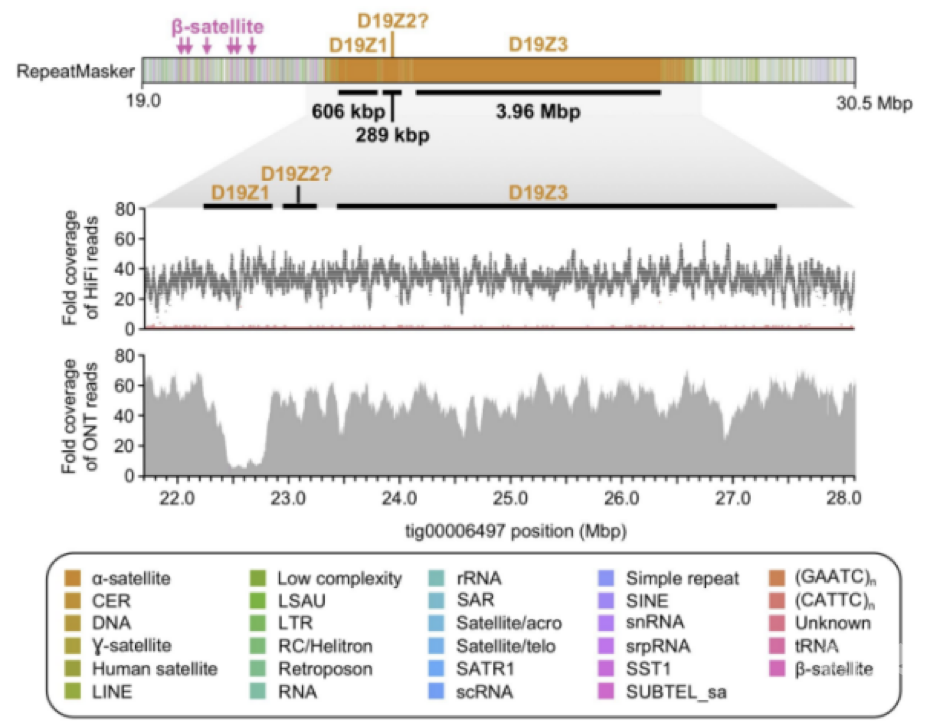
图2 19号染色体着丝粒的组装结果
Ultra-long reads和HiFi reads 强强联合,谁与争锋?
三代测序技术在基因组测序组装领域风生水起,大家难免会把两种测序技术进行比较,Pacbio(PB)公司的Sequel II平台产生的HiFi reads单碱基准确性超过99%; Oxford Nanopore Technologies (ONT)公司的PromethION平台能够产生长达2Mb的ultra-long reads。 HiFi reads 与 ultra-long reads谁是高质量基因组必备技术?用数据说话。
2020年2月,北京希望组、云南生物资源保护与利用国家重点实验与西北工业大学等多家单位共同合作的研究成果“Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacbio Sequel II system and ultralong reads of Oxford Nanopore”在bioRxiv预印发表。本研究分别利用 PB Sequel II和ONT PromethION平台对水稻9311进行测序,将获得的 PB HiFi reads(253 Gb, reads N50=11Kb)和ONT ultra-long reads(92 Gb, reads N50=41Kb)分别用3种软件(Canu1.9, NextDenovo2.0-beta.1, WTDBG2)组装,根据Contig N50选择两种平台的最优组装结果进行比较[4]。结果显示:
在结构水平,ONT ultra-long reads组装的连续性更好,N50达到32M,避免了长重复区域的组装错误,这些错误在PB组装中导致54个注释基因冗余或丢失。由此可见,PB HiFi reads 和 ONT ultra-long reads两种技术组装的基因组各有优势也各有瑕疵。在人类,乃至其他动植物基因组的构建过程中,应该适当调和这两种技术的优势,来减少组装错误,降低对后续注释造成的影响。那么如何将两种技术更好的结合呢?结合之后,组装的基因组能达到什么水平?
2020年7月,Nature杂志上发表的人类X染色体完成图[1],利用Nanopore ultra-long、PacBio、10X Genomics等多种测序技术,成功组装出首个端粒到端粒(Telomere-to-Telomere)的无缺口人类X染色体完成图。这项研究选用CHM13hTERT细胞系,取39×ultra-long数据与70×PacBio数据用Canu软件进行初步组装,获得基因组大小为2.9Gb, 首次完成了从端粒到端粒、完整无缺的人类X染色体序列。本研究中最大的亮点是ONT ultra-long reads和PB数据的应用,提升基因组组装连续性和准确性,跨越上百Kb的片段重复,在重复单元标记引导下跨越2.8Mb的着丝粒区间,以及提供碱基水平染色体范围的DNA甲基化图谱。
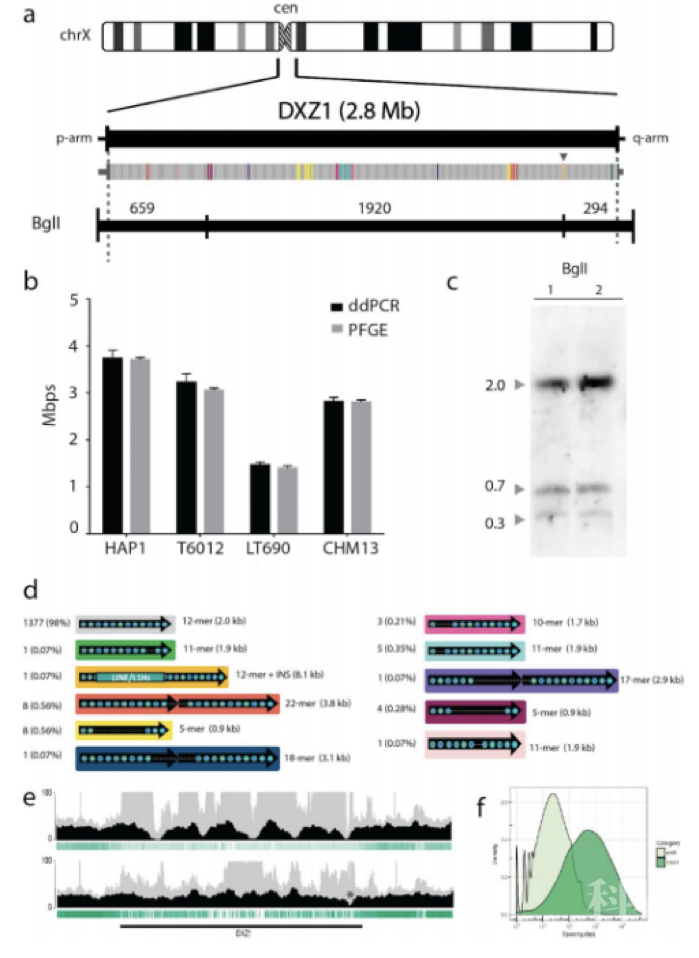
图3 X染色体2.8Mb着丝粒区域验证
动植物基因组近完成图高性价比策略
ONT ultra-long reads+PB HiFi reads+Bionano+HiC
在动植物基因组研究中,已经有很多物种发布了参考基因组,然而这些基因组距离完成图还有一定的距离。基于人类基因组T2T的策略,新方法:ONT ultra-long reads的长读长+PB HiFi reads的高准确性,或许能将动植物基因组提升到一个新水平。希望组持续关注三代技术对基因组组装质量的提升,也是目前国内少数能稳定产出高质量ONT ultra-long reads的服务商之一。自去年8月实现ONT ultra-long单张芯片测序读长N50突破100Kb以来,已经完成了多个不同物种、不同大小基因组的纯ultra-long reads组装。例如,某基因组大小约2G的植物,希望组采用Ultra-long Reads组装contig N50达220.12Mb。
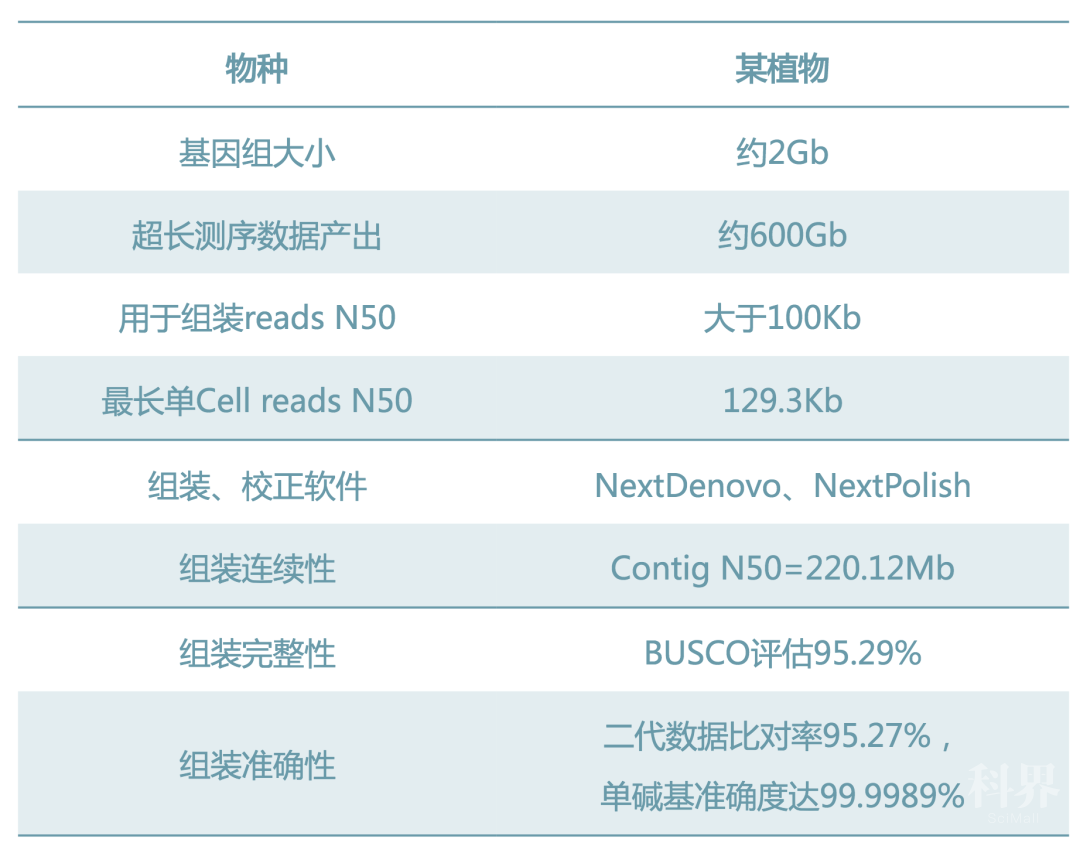
今年,希望组成功引进Sequel II平台,整合ONT和PB双方优势,首发推出基因组近完成图解决方案:ONT ultra-long reads+PB HiFi reads+Bionano+HiC,突破现有基因组技术指标,为客户提供更有效、更完整、更准确的基因组图谱,为后续基因组进化,基因组结构变异,基因功能研究等等更深入的研究奠定基础。
参考文献
[1] Miga K H , Koren S , Rhie A , et al. Telomere-to-telomere assembly of a complete human X chromosome[J]. Nature, 2020.
[2] Jain M , Koren S , Miga K H , et al. Nanopore sequencing and assembly of a human genome with ultra-long reads[J]. Nature Biotechnology, 2018.
[3] Nurk S, Walenz BP, Rhie A, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads[J]. BioRxiv, 2020.
[4] Lang D, Zhang S, et al.Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacbio Sequel II system and ultralong reads of Oxford Nanopore[J]. BioRxiv, 2020.
来源:gh_ef83defd2d1d Nextomics
原文链接:http://mp.weixin.qq.com/s?__biz=MzA5MzI3NDczNQ==&mid=2650986935&idx=1&sn=4035e27253d1bd894c3e73d1a4ed85c6&chksm=8b962eb7bce1a7a113f0fb91cfc285ff7b69f23fed0ea45db523c4a80630f274a37a999d0577&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
新发现填补人类基因组图谱空白

WHO启动全球人类基因组编辑注册计划

《自然》:美国家人类基因组研究所发布人类基因组领域美好愿景!

中国“绿色超级稻”新品种累计推广面积9000万亩

中国人类基因组研究有望“弯道超车”
第一届材料基因组工程高层论坛通知

迄今最全面人类基因组测序完成

人类基因组测序

昆士兰大学统计遗传学家杨剑:耕耘九年,他捧起澳洲最高科学奖

口袋装置测序人类基因组


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号