科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-08-12
来源:X一MOL资讯
目前,耐药性致病菌的层出不穷,已引起全球范围的焦虑和担忧,而抗生素滥用更加剧了这一问题的严重性和紧迫性。国内外许多科研单位和制药公司,都在为获得新型、高效的抗生素而不断探索。为降低来自其他抗生素所致交叉耐药性的影响,研究者们一方面试图寻找新的抗菌作用靶点,另一方面则在设计和筛选具有全新结构的化合物。近年来,多肽作为药物在临床上的应用越来越多,又因其不同于常用抗生素的结构,在抗感染领域受到广泛关注。香港大学李学臣课题组一直专注于环肽类抗生素的化学合成、药物化学和其化学生物学的研究。相继完成了daptomycin(达托霉素)(2013年)、teixobactin(2016年)、A54145(2019年)的首次全合成,以及它们的药物化学研究(J. Med. Chem., 2020, 63, 3161; ACS Med. Chem. Lett., 2020, 11, 1442)。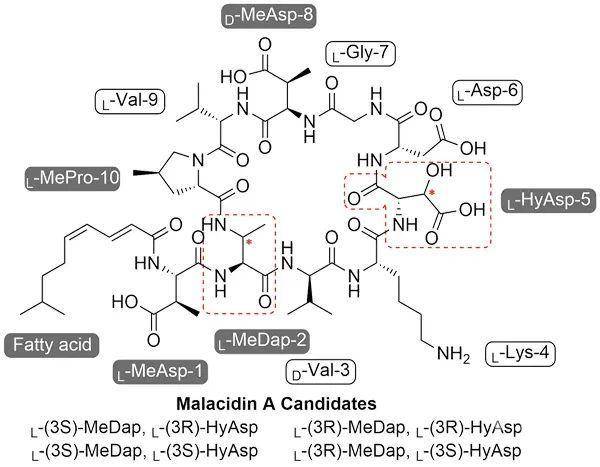
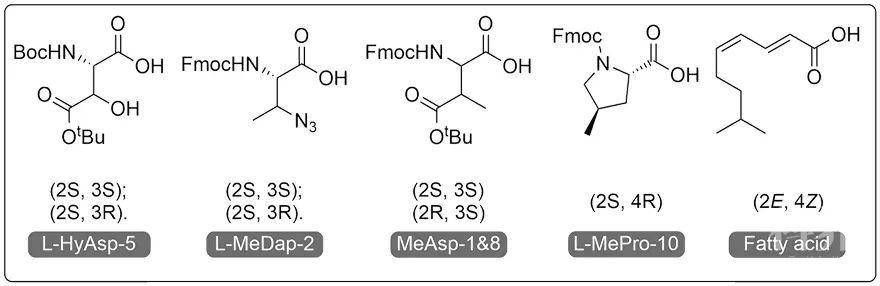
在设计合成了一系列的非天然氨基酸砌块后,作者结合固相多肽合成技术(SPPS),制备了不同的环化前体。接着,作者先是尝试使用液相环化法和树脂上环化法构建Malacidin A结构。由于前者环化效率低、羟基天冬氨酸极易发生侧链内酰胺化和后者前体提前从固相上脱落等原因,最终环化产物无法得到。李学臣教授组早前开发出了一套基于β羟基的丝氨酸/缬氨酸偶联法(Ser/Thr Ligation, STL)(Acc. Chem. Res., 2018, 51, 1643)用于全脱保护多肽之间的化学选择性偶联,已成功应用于多种蛋白质和天然环肽产物的全合成中。作者据此推测,同样带有β羟基官能团的羟基天冬氨酸亦能进行这种偶联反应从而实现高化学选择性的环化。实验证明,羟基天冬氨酸能够与羧基端反应伴侣进行高效偶联,并且没有伴随其他副反应的发生。在这条合成路线上,四种可能的Malacidin A化合物能被高纯度合成。最终,作者通过一维和二维核磁表征和抗生素活性测试对比,确定了β羟基天冬氨酸和3-甲基二氨基丁酸残基的β位点均为R构型。同时,洛克菲勒大学Sean F. Brady教授利用Advanced Marfey分析得到了一致的结论。

来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657653890&idx=3&sn=388226992355f334750faf02a40b55dd&chksm=80f8bd52b78f3444eae0c7bec554a0d48cb157f37bbeb0ee932034e7bad07bf2e639e978a295&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
中国抗癌协会抗癌药物专业委员会召开换届会议



服用抗生素有讲究 一文读懂抗生素的“前世今生”
2016药物/化学品致癌性试验研究高级培训班在青岛举行
科普丨抗生素科普小知识
吃抗生素≠治感冒
人工智能助力新型抗生素研发,缓解抗生素耐药性


抗生素能治疗癌症吗?

让抗生素精准狙击细菌
抗生素你用对了吗?
2015年(第五届)药物毒理学年会在海口市顺利召开





科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号