科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-03-27
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins)是目前最常用的基因编辑工具酶,在科研领域被广泛应用,而在临床方面的应用因为其脱靶活性一度停滞不前。它通过guide RNA(gRNA)与靶向DNA序列的配对,从而将Cas9锚定在靶向基因并诱导产生DNA双链断裂(DSB)。在基因编辑诱发的DSB的修复过程中,一定几率会产生基因突变或者外源DNA片段的插入,从而达到基因编辑的目的【1】。在实际应用过程中,一个好的基因编辑酶Cas9需要同时满足高效切割靶位点、低脱靶活性和低染色体异常三个特点。目前在这三个方面,有一部分基于PCR的方法用于估算基因编辑效率,但其结果可靠性有待提高;有一些基于高通量测序的方法可以在体内或者体外检测基因编辑酶的脱靶活性;尚无系统的可定量的测量基因组异常结构的方法。
2019年3月26号,北京大学生命科学学院和北大-清华生命科学联合中心胡家志课题组在Cell Discovery上发表题为Optimizing genome editing strategy by primer-extension-mediated sequencing 的论文。该研究描述了一种新的方法,可以用来同时定量检测Cas9编辑效率和脱靶活性以及编辑引起的染色体异常结构,即primer-extension-mediated sequencing(PEM-seq,引物延伸介导的测序)。这是对基因编辑和DNA损伤修复等领域都有巨大促进作用的新技术。

依据DNA双链断裂与染色体易位的原理,胡家志课题组在已有的高通量测序方法【2】的基础上开发了灵敏度更高且可以全面且定量评估基因编辑的新方法 PEM-seq。与以往基于二代测序评估Cas9脱靶活性的方法相比,PEM-seq不仅可以灵敏的找出Cas9的脱靶位点,还可以精确定量CRISPR/Cas9在靶向位点的切割效率,从而找到更加高效安全的Cas9切割位点。与此同时,PEM-seq还深度揭示了靶向位点附近由于基因编辑而产生的染色体异常结构,例如大片段缺失、染色体易位等。
以靶向治疗RAG1(Recombination activating gene 1)基因上的RAG1A编辑位点为例,胡家志课题组发现:在Cas9切割位点5kb以内存在着占总编辑事件2.5%的大量的染色体缺失倒位等异常结构;更为严重的是,这些异常结构可以延伸至距离切割位点50kb甚至更长的领域,足以严重威胁基因组的稳定性。这些现象揭示了Cas9应用前评估的必要性。应用PEM-seq可以全面地评估Cas9的切割效率及其所导致的大多异常结构,从而优化基因编辑策略,以达到获得最大编辑效率且尽量减少脱靶活性的效果。
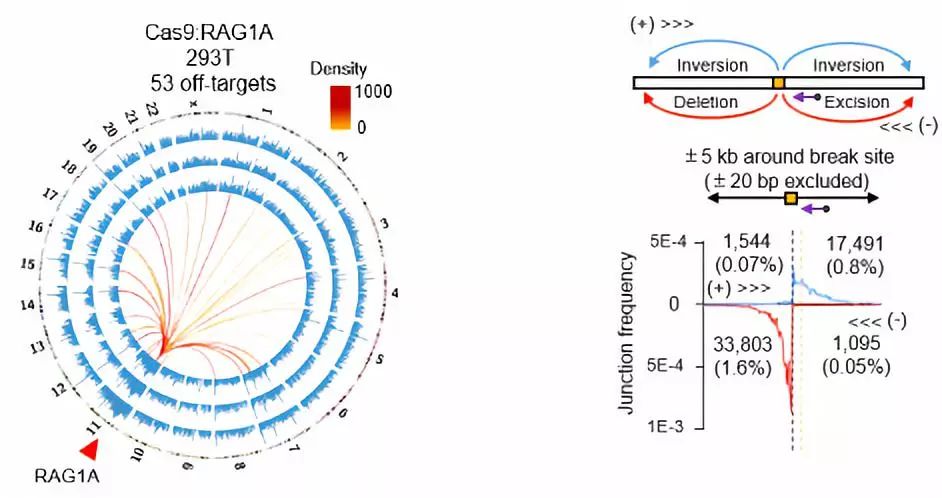
图: PEM-seq检测出Cas9 在RAG1A位点有53个脱靶位点(左);PEM-seq检测到在Cas9切割位点5kb内存在着大量染色体异常机构(右)
同时,为了降低CRISPR/Cas9的脱靶活性,胡家志课题组将现有Cas9变体的突变位点进行了组合筛选,通过PEM-seq筛选出了一个切割效率与野生型Cas9相当但脱靶频率明显更低的变体FeCas9。该酶的使用方法与传统Cas9类似。
此外,PEM-seq在基因组的稳定性研究方面还有较大的潜力。基因组的修复与DNA修复等因素密切相关,而传统的方法多用PCR或者分子克隆获得修复信息,样本小且存在偏好性。PEM-seq与一些之前的高通量测序方法类似,可以提供较大的数据量,且相对而言有一个更加明显的优势,即定量。PEM-seq可以对DNA修复的步骤进行逐步定量,从而细致地描画DNA从损伤到修复的过程,以获得更加精准的模型。
据悉,胡家志课题组的博士生尹健行,博士生刘孟竺和博士后刘阳为该文章的第一作者。
原文链接:
https://www.nature.com/articles/s41421-019-0088-8
制版人:半夏
参考文献
1. Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases.Nat. Biotechnol. 31, 827–832 (2013).
2. Hu, J. et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 11, 853–871 (2016).
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652468061&idx=5&sn=8484c1c0556307dc0acf0c0f75bd6804&chksm=84e2e0e9b39569fff2a238254440677b9867bc5bbc1a6d62bc7378b759edc256c9ab5eb68f45&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
【大师讲堂】神奇的DNA损伤修复
细胞修复DNA损伤新机制揭示
DNA修复之谜
王艳丽:做研究就像破案
NCB:相分离可调控DNA损伤修复

黄俊:修复“生命密码”——DNA
演化:“最奇怪动物”身份揭秘
合成生物学: 一个用来控制转基因生物的内置毁灭开关

水稻DNA双链断裂修复基因克隆成功
毛志勇组建立新的DNA修复检测系统并阐述染色质环境下DNA修复调控机制


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号