科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-04-04
来源:iNature
2019年4月2号,深圳华大基因研究院在Genome Research上在线发表了题为Efficient and unique co-barcoding of second-generation sequencing reads from long DNA molecules enabling cost effective and accurate sequencing, haplotyping, and de novo assembly的文章。在这里研究人员开发了stLFR技术,这是一种利用经济的第二代测序技术从长DNA分子中获得序列数据的技术。stLFR代表了一种易于自动化的解决方案,可实现高质量的测序,定相,SV检测,支架,经济高效的二倍体从头基因组装配以及其他长DNA测序应用。


到目前为止,绝大多数个体高等生物体全基因组序列缺乏关于单碱基到多碱基变异在同源染色体上作为连续传递的顺序信息,通常称为单倍型。此外,大多数测序的基因组留下了未解决的新序列,在参考基因组、较大的结构变异和其他难以用现有技术进行分析的区域中找不到。对于许多早期的基因组研究来说,这一信息并不重要,而且被忽视了。然而,当我们更全面地理解一个人的基因组如何对他们所表现出的无数表型做出贡献时,这个缺失的信息就变得必要了。
最近开发了许多技术,包括直接单分子测序,以产生至少一部分这种信息。大多数是基于共条码的过程,即在单个长基因组DNA分子的子片段中加入相同的条形码。测序后,条形码信息可以用来确定哪些读取是从原始的长DNA分子中提取出来的。这一过程首先由Drmanac描述,并由Peters等人用384孔板测定。这些方法在技术上很难实现,价格昂贵,数据质量低,不单独分析单个DNA分子(即,不提供独特的协同条形码),或将这四种方法结合在一起。在实践中,大多数都需要一个单独的全基因组序列生成的标准方法,以改善变异调用。
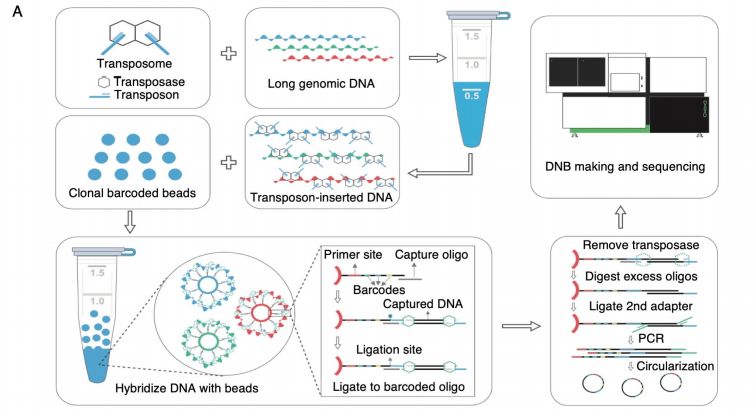
在这里,研究人员描述了一种名为stLFR的技术,这是一种有效的DNA协同条形码方法,在单个管中启用了数百万个条形码。这通过使用微珠的表面作为隔室(例如,384孔板的孔)的替代物来实现。每个珠子携带许多拷贝的独特条形码序列,该序列被转移到每个长DNA的亚片段上。该序列于2019年4月3日发表 - 由Cold Spring Harbor Laboratory Press分子出版。然后在常见的第二代测序组装(例如BGISEQ-500,MGI-2000或等同物)上分析这些共同条形码的子片段。
原文链接:
https://genome.cshlp.org/content/early/2019/04/02/gr.245126.118
来源:Plant_ihuman iNature
原文链接:http://mp.weixin.qq.com/s?__biz=MzU3MTE3MjUyOA==&mid=2247500069&idx=6&sn=9793c95a7724785502afc26edbaa4caf&chksm=fce6b2facb913bec850766f0a71f6a0b7d14b61edffff025946f8295da9488a99f0c81dad44f&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
DNA指纹分析也有漏洞?科学家引入DNA条形码
光学条形码阅读器

昆明动物所等发布中国蛇类DNA条形码参考数据集

DNA“条形码”瞄准生物大发现
王艳丽:做研究就像破案
DNA条形码揭示癌细胞逃避免疫能力

新研究揭开人类头发曲直的秘密

夫妻相真的存在,研究发现夫妻DNA相似度高过陌生人

DNA到底能不能预测外貌?
条形码中的数学奥秘——二进制


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号