科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-07-30
来源:BioArt
ybt :NCB | 楼振昆团队揭示AMPK-Parkin通过抑制坏死小体负向调控坏死和肿瘤生成的机制
Necroptosis是程序性坏死或者炎症性细胞死亡,关联着炎症反应和肿瘤发生、进展。RIPK3是程序性坏死的起始因子,RIPK3磷酸化促进RIPK3和RIPK1形成坏死小体(necrosome)【1】。随后,MLKL被招募并活化,诱发炎症反应【2】。但是,坏死小体如何被负向调控目前尚不清楚。
AMPK是高度保守的丝氨酸/苏氨酸蛋白激酶,胞内的ATP含量偏低和AMP水平上升导致AMPK激活【3】。AMPK能监测并维持能量的稳态平衡。之前的研究表明AMPK具有抗炎或抗肿瘤功能。程序性坏死和细胞内ATP缺乏相关,AMPK和程序性坏死之间或许存在关联。
Parkin是E3泛素连接酶,它的失活在青少年帕金森病和多种人类肿瘤中常见【4,5】。Parkin敲除的小鼠对炎症相关的退行性疾病易感【6】,显示出抗炎作用;Parkin杂合缺失促进小鼠结肠癌的进展,显示出抗肿瘤功能。但Parkin如何实现抗炎、抗肿瘤功能还有待研究。Parkin、AMPK、坏死小体三者之间是否存在关联,如何行使功能目前尚不清楚。
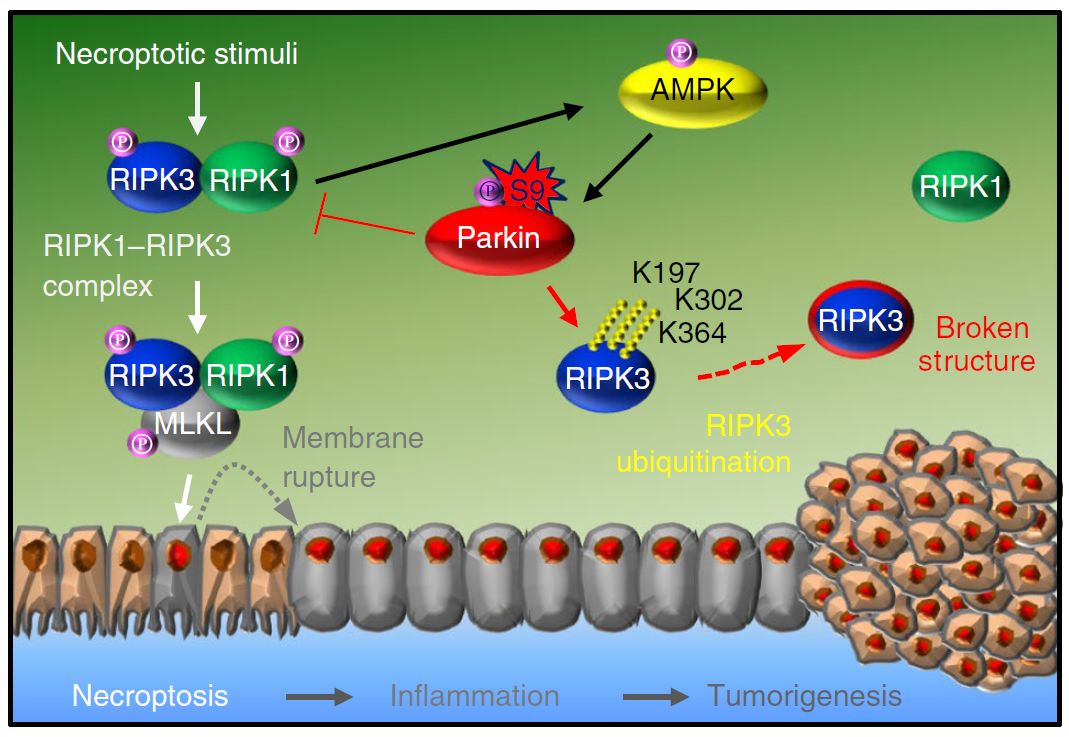
2019年7月29日,来自温州医学院的 Jin-San Zhang 和美国的Zhenkun Lou团队合作在Nature Cell Biology上发表了题为The AMPK–Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome的文章,揭示了坏死过程,AMPK磷酸化并激活E3泛素连接酶Parkin,Parkin通过促进RIPK3多泛素化抑制RIPK1-RIPK3形成坏死小体,负向调控程序性坏死和炎症。证明了AMPK-Parkin是调控程序性坏死和炎症的重要机制。

研究团队首先从Parkin的生理功能入手,发现Parkin KO小鼠在10月龄出现小肠部位炎症性细胞浸润、体重下降、脱肛、脾脏肿大等炎症增加及初步出现增生现象。研究人员提出假设:Parkin敲除导致炎症增加,进而诱导肿瘤发生和进展。Parkin KO上调促炎细胞因子TNF- α、IL-1 β和IL-6在小肠细胞和小鼠胚胎成纤维细胞(MEFs)中表达;野生型小鼠在12月龄不能产生肠息肉,84%的Parkin KO小鼠则出现5-10个肠息肉。对溃疡性结肠炎的病人样本进行检测,发现发炎的升结肠或降结肠组织中Parkin的表达下调。这些结果初步显示Parkin具有抗炎功能。

Parkin如何行使抗炎功能?T/C/Z(TNF/cycloheximide/z-VAD)处理导致Parkin KO细胞进行程序性坏死,而不是凋亡或自噬,Parkin KO导致RIPK1/RIPK3、MLKL的磷酸化增加。免疫共沉淀实验显示,T/C/Z处理导致内源的Parkin和RIPK3结合,而不是RIPK1或者MLKL,而且Parkin的IBR、R2结构域和RIPK3的N端结构域对两者的结合是必需的。Parkin KO MEFs中表达Parkin导致RIPK1-RIPK3复合体形成减少。在IBR-R2结构域中突变的R420H、H461R导致Parkin与RIPK3的作用减少,对RIPK3的磷酸化和程序性坏死抑制作用消失;E3连接酶活性结构域突变的K161N、T240R虽然能够于RIPK3正常作用,但是不能抑制程序性坏死。进一步研究发现,Parkin促进RIPK3进行K33连接的多泛素化修饰,修饰的位点为K197、K302、K364,多泛素化的修饰抑制RIPK3形成puncta-like的亚细胞定位,抑制坏死小体的形成。所以,T/C/Z处理下,Parkin和RIPK3结合并对其进行多泛素化修饰,阻止RIPK3与RIPK1形成坏死小体,抑制程序性坏死。
那么程序性坏死过程中,Parkin如何被激活?之前的研究显示AMPK KO细胞对T/C/Z诱导的细胞死亡更多敏感,而细胞内ATP耗竭是程序性坏死的主要特征之一,ATP耗竭是激活AMPK的重要信号。研究发现,AMPK的起始活化依赖于RIPK,AMPK的持续活化则是由MLKL的激活或者细胞内的低ATP水平造成的。Parkin的S9残基被预测为AMPK的磷酸化位点,其周围的序列与保守的AMPK磷酸化motif相符,研究人员猜测Parkin是AMPK的底物。体外试验表明,AMPK可以直接磷酸化Parkin的S9位点。而AMPK缺失抑制程序性坏死过程中Parkin的磷酸化,RIPK3的泛素化降低;突变Parkin的S9导致其无法被AMPK磷酸化,而且RIPK3磷酸化增加。故程序性坏死中,AMPK介导Parkin磷酸化,激活Parkin。
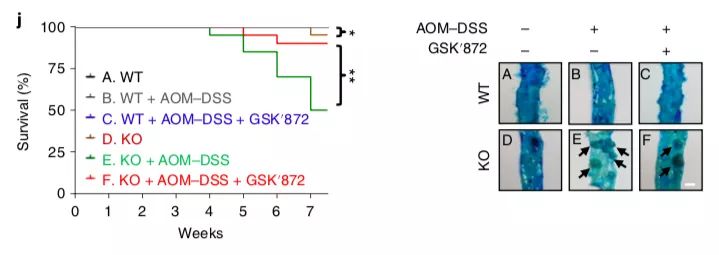
最后,失调的程序性坏死是否参与Parkin KO小鼠的肿瘤发生?RIPK3的抑制剂GSK’872显著降低Parkin KO小鼠中RIPK3的磷酸化、肠息肉发生,增加生存率,说明RIPK3的过度激活和程序性死亡与Parkin KO小鼠的肿瘤发生相关。GSK’872处理显著降低Parkin KO小鼠中促炎细胞因子TNF- α、IL-1β和IL-6表达,显示出RIPK3的过度活化导致Parkin KO小鼠中的炎症增加。同时,AOM-DSS诱导的IBD模型中,Parkin KO导致RIPK3磷酸化、促炎细胞因子、肠息肉数目增加,小鼠生存下降;GSK’872处理后则显著降低RIPK3磷酸化、促炎细胞因子、肠息肉数目;AMPK的激活剂metformin和A-769662显著抑制野生型小鼠中AOM-DSS诱导的肠息肉生长,在Parkin KO小鼠中没有作用。对健康人和IBD病人的结肠组织进行检测,发现Parkin的表达和RIPK3的磷酸化呈负相关。
总之,本项研究揭示了AMPK-Parkin是程序性坏死和炎症反应的重要调控因子,其失调导致炎症相关的肿瘤发生,为研究程序性坏死的负向调控提供了新的视角。
原文链接:
https://doi.org/10.1038/s41556-019-0356-8
参考文献
1. Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
2. Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
3. Kemp, B. E. et al. AMP-activated protein kinase, super metabolic regulator. Biochem. Soc. Trans. 31, 162–168 (2003).
4. Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 (2000).
5. Fujiwara, M. et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27, 6002–6011 (2008).
6. Thomas, M. Inflammation in Parkinson’s Disease (Springer, 2014).
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652473299&idx=2&sn=edbe43b6493fb621230dc62772d81d85&chksm=84e21467b3959d7131a36b90d9c3fc0ff43767b91dc575782bd81d2fecf74d453cf186c81f77&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
发现新型磷酸化蛋白识别机制

磷酸化调控蛋白质结合机制研究取得进展

福建农林大学熊延课题组揭示了调控植物生长的 “磷酸化密码”
研究揭示氢离子焦磷酸化酶调控甘薯品质和产量的作用机理

磷酸化修饰组学揭示KRAS突变肿瘤的精准治疗新策略

【大师讲堂】诺奖得主Edmond H. Fischer:磷酸化与去磷酸化调控的生物网络

Nat Biotech:“机器学习”帮助鉴定磷酸化位点
光合磷酸化作用
研究揭示磷酸化修饰调控内质网应激早期应答新机制
开发出酪氨酸磷酸化监测新方法


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号