科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-10-13
来源:宏基因组

Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference
Nature Methods [IF:28.467]
2017-03-06 Brief Communication (短通讯)
DOI: https://doi.org/10.1038/nmeth.4197
第一作者:Rob Patro1
通讯作者:Rob Patro1 和 Carl Kingsford5
其它作者:Geet Duggal2, Michael I Love3,4,
Rafael A Irizarry3,4
作者单位:
1美国纽约州石溪市,石溪大学计算机科学系(Department of Computer Science, Stony Brook University, Stony Brook, New York, USA)
5美国宾夕法尼亚州匹兹堡,卡内基梅隆大学计算生物学系(Computational Biology Department, Carnegie Mellon University, Pittsburgh, Pennsylvania, USA)
导读Salmon可提供快速且偏见的转录表达定量
Salmon是一种准确快速定量转录本丰度的方法;
它是第一个可校正转录组片段GC含量范围内偏差的定量工具,大大提高了丰度估计的准确性以及后续差异表达分析的可靠性;
新的双相并行推理算法和功能丰富的偏差模型与超快速读长映射过程结合在一起;
其计算速度快,硬盘空间占用少等优点,也在宏基因组基因定量中广泛应用;
软件支持多种常用安装方式,且支持多样本汇总为表格。
点评:传统的定量工具采用比对的方法,对于人类几万个基因都需要消耗几小时,而面对微生物组动辄千万的基因更是费时费力。Salmon的非比策略具有速度上的极大优势,同时不会生成传播的巨型SAM格式比对文件。Salmon又是体现了出版平台对文章引用存在巨大影响的例子,该软件2015年发布在BioRxiv上面,两年只有10来次的引用,截止目前4年也只有37个引用。而文章被Nature Methods接收发表后,短短两年引用达891次,相同时间内此用高达上百倍,可见这咱28分的杂志确实获得了同行的广泛关注。所以好的东西,传播平台也很重要,比如发了文章记得来宏基因组公众号宣传,让你免费体验高分文章才有的传播效果。

我们介绍Salmon,这是一种从RNA-seq读长中定量转录本丰度的方法,该方法准确而又快速。Salmon是第一个可校正转录组片段GC含量范围内偏差的定量工具,我们证明该方法大大提高了丰度估计的准确性以及后续差异表达分析的可靠性。Salmon将新的双相并行推理算法和功能丰富的偏差模型与超快速读长映射过程结合在一起。
详者注:目前该软件在宏基因组上千万数量的基因定量中有广泛应用,效率惊人。
主要结果使用来自GEUVADIS和SEQC研究的实验数据以及来自Polyester和RSEM-sim仿真器的综合数据,我们将Salmon与Kallisto和eXpress + Bowtie2进行了基准比较, 这两种方法也都实现了自己的偏差模型。我们还使用传统比对(来自Bowtie2)作为输入(表示为“Salmon(a)”)测试了Salmon。我们显示Salmon在准确性方面通常胜过Kallisto和eXpress(图1以及补充图2和3)。我们注意到,所有这些工具都解决了转录本量化问题,并且没有识别或组装新颖的转录本(补充说明1)。
图1. Salmon与同类软件对比的表现Figure 1 | Performance of Salmon
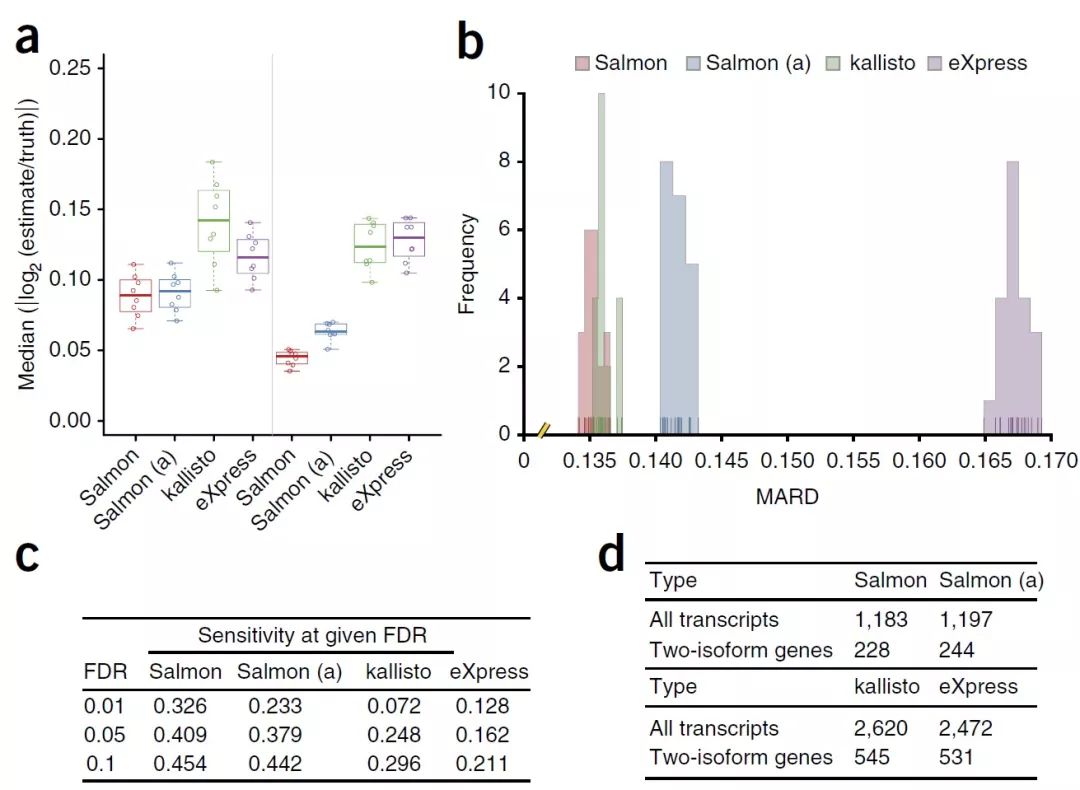
(a)在聚酯(Polyester)模拟数据的所有16个重复下,估计丰度和真实丰度之间的绝对对数倍数变化的中位数(log-transformed fold changes,lfc)。lfc越接近0,真实和估计的丰度就越相似。左面板和右面板显示了使用从实验数据(在线方法中的详细信息,地面真实情况模拟数据)获悉的不同GC偏向曲线模拟的样品下,对数倍数变化的分布。
(b)Salmon与传统比对方法kallisto和eXpress的比较,准确度指标中所述的平均绝对相对差异(mean absolute relative differences,MARD)的分布,该结果由RSEM-sim生成了20个模拟重复。Salmon和Kallisto产生的MARD相似,尽管Salmon的MARD分布比Kallisto的小得多(Mann-Whitney U检验,p = 0.00017)。两种方法都优于eXpress(Mann-Whitney U检验,p = 3.39781×10-8)。
(c)在典型的FDR值下,使用Salmon的估计找到真正的差异转录本的敏感性比使用Kallisto的估计要高53%–450%,比使用eXpress的Polyester模拟数据的估计要高210%-250%。
(d)对于30个GEUVADIS样本,当组之间的对比度仅是技术上的混淆(即测序的中心)时,预期FDR为1%的转录本数量称为差异表达(differentially expressed,DE)。Salmon产生的DE转录本少于其他方法的一半。对样品进行置换,或在测序中心内测试DE,所有方法的DR<1%转录本平均称为DE。
来源:meta-genome 宏基因组
原文链接:http://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247488955&idx=1&sn=5ae58a0e177a9a932391dcc97933c1f4&chksm=fab9ff0acdce761cc2099899f5fc0d18f43f3074e95526bee7b06735829a7a7f05b810a98114&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

蒲慕明院士:建设世界一流期刊关系学术创新的发言权

新突破! 二维实验系统成功应用于四维材料研究
Nature子刊:Salmon不比对快速宏基因组基因定量
华侨大学博士生首次在Nature发文



宏基因组理论教程2扩增子分析

Nature综述:2万字带你系统入门鸟枪法宏基因组实验和分析

宏基因组如何指导微生物分离培养

PICRUSt2预测宏基因组功能
Nature综述:临床宏基因组学的应用与挑战
宏基因组公共数据挖掘基因组集再发Nature


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号