科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-10-29
来源:研之成理

▲第一作者: 葛亮 ;通讯作者:冯超教授,Patrick J. Walsh 教授
通讯单位: 南京工业大学,宾夕法尼亚大学
论文DOI:doi.org/10.1038/s41467-019-12403-2
全文速览
冯超教授课题组与宾夕法尼亚大学 Patrick J. Walsh 教授课题组合作发展了一种基于芳基自由基正离子活化和亲核试剂进攻协助的非活化芳基环丙烷的选择性开环胺氧化反应新策略。此外,作者还发现以简单烯烃为起始原料,通过连续环丙烷化-胺氧化能够实现β-胺基酮类化合物的快速制备,而且该反应可以顺利放大到克级规模。
背景介绍
环丙烷作为一类重要的有机结构单元广泛存在于天然产物以及各种药物活性分子中,同时,三元环结构较大的张力使得此类化合物可以经开环官能反应制备结构多样的有机骨架,这也使得环丙烷类化合物成为现代有机化学中重要的合成砌块。
目前环丙烷类化合物的开环策略主要包括:
1)以三元环环上邻近碳原子分别含有供电子基团和吸电子基团的环丙烷类化合物为起始原料,经路易斯酸活化后三元环会发生碳-碳键断裂的官能化反应,然而这种策略对应的底物适用范围受到很大限制;
2)利用三元环对低价过渡金属的直接氧化加成,继而发生开环反应,或者是利用过渡金属催化中的 β-碳消除策略,从而实现碳-碳键断裂开环反应,但是这些方法通常需要特殊的导向基团或者螯和官能团来确保碳-碳键断裂反应的位点选择性;
3)通过强路易斯酸直接活化三元环从而实现碳-碳键断裂开环,但是适用于该方法的亲核试剂种类较少,限制了该策略在有机合成中的实际应用。
随着可见光促进的光氧化还原催化的不断发展,Nicewicz 课题组于 2014 年报道了光催化烯烃的反马氏氢胺化反应。经单电子氧化产生的烯基自由基正离子中间体可以与胺类化合物发生亲核加成,从而实现烯烃的氢胺化反应。受此启发,作者课题组在 2019 年初利用单电子氧化活化惰性官能团的策略成功实现了富电子偕二氟烯烃的氟烯丙基化反应()。基于此作者设想利用该策略实现非活化芳基环丙烷的选择性开环/官能化反应。
本文亮点
南京工业大学冯超教授课题组报道了可见光光氧化还原催化的芳基环丙烷的开环胺氧化反应,为 β-胺基酮类化合物的构建提供了一种新策略。在可见光照射下,芳基环丙烷经激发态光催化剂的单电子氧化可生成芳基自由基正离子中间体,进而活化环丙烷骨架,然后该中间体在亲核试剂进攻下能够实现选择性开环及后续的官能化反应。
该策略的特点在于:
1)通过原位形成的芳基自由基正离子活化惰性环丙烷;
2)氧气在反应中扮演双重角色,既充当氧化剂使光催化剂再生,又作为反应底物氧化苄基自由基;
3)反应生成的β-胺基酮类化合物不仅是很多天然产物的核心结构,而且还是有机化学中的重要合成砌块;
4)此外,作者还发展了一种以简单烯烃为起始原料,通过连续环丙烷化-胺氧化快速构建β-胺基酮类化合物。
图文解析
在最优反应条件下,作者首先对芳基环丙烷进行了底物拓展,含有电中性基团以及给电子基团的芳基环丙烷普遍适用于该反应,均能以较高的产率得到对应的目标产物。此外,作者还对氮亲核试剂的范围进行了探究,发现吡唑类、三氮唑类以及三氟甲磺酰胺经反应都能以中等至较优收率得到目标产物(图1)。

▲图1. 底物拓展
接下来作者又成功地将 Yoshida 的环丙烷化与开环胺氧化策略有机结合,以简单烯烃为起始原料,通过连续环丙烷化-胺氧化反应,实现了β-胺基酮类化合物的合成以及克级规模反应(图2)。
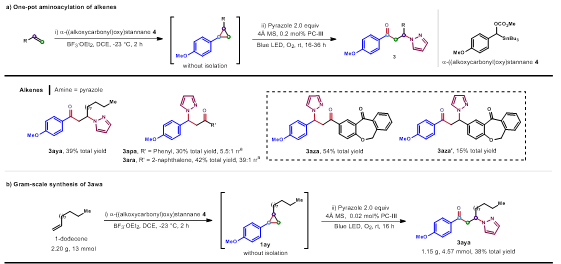
▲图2. 烯烃一锅法反应及克级实验
为了探究反应的历程,作者开展了一系列控制实验。首先,在标准反应条件下,向环丙烷 1b 和吡唑 2a 的反应中加入自由基抑制剂 TEMPO 后,发现未能检测到目标产物 3ba 的生成,这表明反应可能有自由基中间体参与(图3 a)。
此外,烷基过氧化氢中间体的分离进一步表明了反应过程中苄基自由基的生成。作者推测氧气分子在捕捉苄基自由基后形成的烷基过氧化氢中间体经脱水作用即可生成目标化合物(图3 b)。为了验证该光氧化还原反应是如何启动的,作者开展了 Stern–Volmer 荧光淬灭研究,研究结果表明只有环丙烷底物能够淬灭激发态的光催化剂(图3 c)。
通过开展手性底物参与反应的研究,作者发现光学纯度较高的原料经过反应后得到光学纯度降低的产物,同时原料自身在光照条件下也会发生消旋化,因此消旋化产物的生成主要是由原料消旋化导致的。据此,作者推测原料胺氧化过程主要是通过协同亲核进攻/开环(SN2)机制来进行的,而非环丙烷先开环得到正离子中间体再接受亲核进攻(图3 d)。
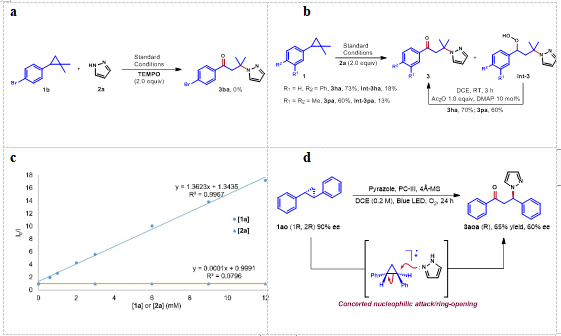
▲图3. 机理研究。a TEMPO 抑制反应;b 过氧化氢中间体的产生及其向目标产物的转化;c 1a 和 2a 的Stern–Volmer 荧光淬灭研究;d 手性底物的相关实验。
基于对照实验与循环伏安实验的结果,作者提出了该反应可能的机理(图4)。首先,IrIII 光催化剂被蓝光激发形成的激发态 *IrIII,随后被芳基环丙烷 1 还原淬灭形成还原态 IrII,并伴随着芳基环丙烷自由基正离子中间体 I 的产生。
与此同时,亲核试剂选择性进攻芳基环丙烷自由基正离子中间体I的富电子烷基取代位点,导致中间体 I 开环生成自由基中间体 II,随后自由基中间体 II 被氧气捕捉生成烷基过氧基自由基 III。烷基过氧基自由基 III 与还原态光催化剂 IrII之间经单电子转移过程生成烷基过氧化氢中间体 IV,同时将还原态光催化剂 IrII 氧化为基态的 IrIII 光催化剂,完成催化循环。最后烷基过氧醇中间体 IV 脱水生成 β-胺基酮类化合物 3。
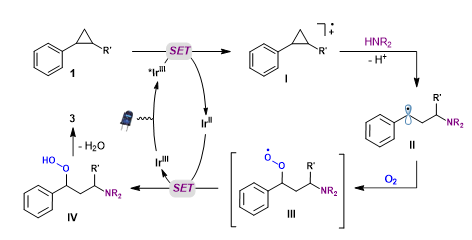
▲图4. 反应机理
总结与展望
综上,冯超教授课题组发展了一种基于芳基自由基正离子活化和亲核试剂进攻协助的非活化芳基环丙烷的选择性开环胺氧化反应新策略。以简单烯烃为起始原料,通过连续环丙烷化-胺氧化实现了 β-胺基基酮类化合物的快速合成,并完成了克级规模反应的尝试。鉴于该反应底物范围广、实用性强,因此其有望在新药先导化合物的发现中得到更多应用。
来源:rationalscience 研之成理
原文链接:http://mp.weixin.qq.com/s?__biz=MzIwMzE5MzQ1NQ==&mid=2649335343&idx=5&sn=629649ef004bc75aab0b1080b5073c3c&chksm=8ece2b2fb9b9a2396f9d93950acf0619c48c8b011fab7a512091f9cc3d266944caf9dd3cd394&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
Armido Studer课题组报道苯炔作为自由基受体实现自由基的串联反应

芳基二价铜化合物的自由基反应、催化及机理研究取得进展
氮杂环卡宾催化醛和氧化还原活性的酯的脱羧烷基化反应

铬催化烯丙基脱氟反应实现羰基自由基与烯烃的偶联

Mn介导的原子转移反应产生氮中心自由基及其应用研究
血红素生物合成途径中SAM自由基酶HemN机制研究新突破
串联自由基环化/分子间偶联反应合成苯并呋喃衍生物
【催化】意料之外的催化活性:硼自由基让氮化硼活泼起来

碘催化C(sp3)-H键的分子间自由基胺化反应
Angew:NHC催化实现烯烃的自由基酰基氟烷基化反应


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号