科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-11-20
来源:研之成理

利用可持续的电能将氮气转换成氨为制造高附加值化学品提供了一条绿色环保的道路。为了进一步提高电催化合成氨的效率,目前还需要在电催化剂开发和电解池设计方面取得进一步进展。在这项研究中,作者通过合理设计电催化剂和电解槽达到同时调节电催化氮还原反应(ENRR)的化学动力学和热力学过程。本文利用石墨双炔(GDY)为基底,利用石墨双炔独特的孔结构来限域金属单原子(Rh,Ru,Co),达到高负载量的金属单原子,并实现了在高压氮气还原过程中ENRR活性的增强。值得注意的是,在加压环境下,金属单原子催化剂的高活性析氢反应被有效抑制,而所需的合成氨反应活性却得到提高。结果表明,在施加55个大气压下,Rh金属单原子催化剂的NH3转换率为74.15 μg h-1 cm-2,FE为20.36%,NH3分电流为0.35 mA cm-2,相比于室温条件下分别显示有7.3倍、4.9倍和9.2倍的提高。此外,使用净化后的同位素15N2气体电解实验证实了氨的氮气到氨的转换。在这项工作中,催化剂和电化学装置的协同作用为工业化电化学合成氨开辟了可能。
背景介绍
氨是人类繁衍和繁荣昌盛所必需的化学品。当今全球氨的生产过度依赖于Harber-Borch法,即在高温(300-500 ℃)和高压(200-300 atm)下将高纯的氮气和氢气转化为氨。迄今为止,这一百年历史的合成氨方法已经为氨的产量做出了巨大贡献,但是也伴随着巨大的全球能源消耗和显著的温室气体排放。电催化N2还原反应(ENRR)可以在温和条件下利用可持续的清洁能源电能将氮气和水合成氨,但目前为止其效率还很低。
电催化合成氨的主要障碍在于N2分子的固有惰性、高的活化能垒、多电子质子转移的反应过程、N2分子在水溶液中的低溶解度以及强的竞争性析氢反应(HER)。解决上述问题需要同时调整反应过程的动力学和热力学过程,从而降低竞争的析氢反应,提高氨的转换效率。从动力学角度来看,许多以催化剂开发为中心的策略,如引入合金、缺陷、掺杂和应变等,已被探索以改善氮还原性能和降低析氢。然而,总体ENRR效率仍然有待提高。另一方面,调节合成氨反应的热力学过程,例如调节电化学反应条件(加压),可以提供同样积极的效果,也可以有效地促进N2还原过程和抑制析氢反应,而这一领域在过去很少被研究。在近年来的研究中,对电催化剂或电解池器件创新的探索经常是独立开展的,尽管它们之间存在着不可分割的相互联系。事实上,ENRR朝着预想的实际应用的方向发展,在很大程度上取决于电催化剂和电化学电解池装置的合作开发。
基于此,作者通过合理设计电催化剂和电解槽达到同时调节电催化氮还原反应的化学动力学和热力学过程。
图文解析

▲图1. (a)不同金属单原子(Rh、Ru、Co)负载石墨双炔的合成示意图;(b、c、h、i、n、o)金属单原子的HAADF-STEM图;(d、e、f、g、j、k、l、m、p、q、r、s)对应金属单原子的EDX-mapping图;图中比例尺为5 nm;
A、 材料合成
M-SA/GDY制备的合成原理是建立在C-C偶联的还原消除反应的基础上的。通常,在还原消除步骤中,底物和金属阳离子形成配位络合物,接着金属中心接受来自底物的电子,从而形成C-C键以及低价金属中心。本项研究采用这种反应机理,设计了一种高效的一锅法合成工艺(图1a),用于制备高载量的金属单原子负载的石墨双炔材料。其反应过程如下:在吡啶作用下,石墨双炔(GDY)单体经一定程度的脱质子化,与溶液中的Rh3+、Ru3+和Co3+形成配位络合物,然后GDY单体在高温下发生C-C偶联反应。通过所形成的石墨双炔空间限域将还原金属原子锚定在GDY骨架上,从而得到高载量的金属单原子负载石墨双炔材料。从图1a中的球差电镜中可以看到明显的密集的金属单原子的负载,而对应的EDX-mapping也证实了金属元素及C元素的均匀分散。
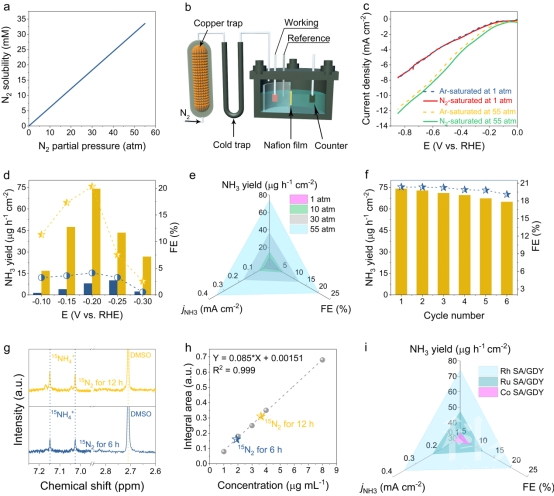
▲图2. (a)氮气溶解度与分压的关系图。(b)加压ENRR装置示意图。(c)Rh-SA/GDY在室温和55 atm条件下的Ar饱和和N2饱和LSV曲线。(d)Rh-SA/GDY在室温和55 atm条件下的NH3产率及其相应的FE。(e)Rh-SA/GDY的NH3产率、FE和jNH3与施加N2压力的对比。(f)Rh-SA/GDY在-0.2 V下的循环稳定性测试(g,h)同位素15N2在6h和12h的电还原反应的1H-NMR谱及对应15NH4+产率。(i)制备的Rh,Ru,Co-SA/GDY的最佳NH3产率、FE和jNH3的比较。
B、加压电催化氮还原性能测试
在室温条件下(25oC和1atm)氮气在水中的溶解度很差,氮气在水中的浓度为6.1×10-4 M,这种低浓度的N2伴随着其缓慢的动力学,导致了低N2的吸附和活化。根据Henry定律,水中溶解的氮气浓度与氮气分压成正比(图2a)。在较高的氮气分压下,预计更多的氮气可以被输送到电极表面。这项工作中在膜分离的三电极电池中考察了ENRR在室温和加压条件下的氮还原的性能指标。在这两种条件下,高纯度氮气(99.999%)依次通过氮氧化物杂质捕集器和液氮冷却捕集器进行净化,以捕获气体中氮氧化物和残余NH3等杂质。不同的是,在室温条件下,纯化后的14N2/15N2气体在电解槽中连续循环,而在加压条件下,净化的14N2气体在直接鼓入高压反应釜中(图2b)。这两种的装置不仅有助于消除外来的氮氧化物污染,而且有助于减少供应气体的消耗,尤其是昂贵的15N2气体,因此,可以得到一个准确且经济有效的电解方案。应注意的是,加压电解槽中的阳极室不仅含有0.005 M H2SO4和0.1 M K2SO4的混合溶液,而且还含有额外的0.01 M抗坏血酸,以防止析氧反应。
以Rh-SA/GDY为代表,在55 atm的压力下,在N2-和Ar饱和的电解液中测试的LSV曲线显示出比室温条件下更明显的电流间隙,这可能是由于更高浓度的N2有利于ENRR的反应所致(图2c)。接下来,在不同的压力下,选择不同电位,对Rh-SA/GDY进行了计时电流测量,以考察NH3的产率和法拉第效率(FE)。对电解后的电解液进行离子色谱分析,可以发现明显的NH4+信号,保留时间约为4 min。在1 atm下,Rh-SA/GDY表现出较平庸的ENRR活性,并且随着电位负移,相应的NH3产率和FE逐渐增加,在-0.25 V和-0.2 V下分别达到10.10μg h-1 cm-2和4.12%的峰值。继续增加电位,由于强的析氢导致N2在电极表面的扩散限制,其相对性能下降。将电解槽中的氮气压力提高到10 atm、30 atm和55 atm,可显著提高NH4+的产量,其对应的最佳性能如下:10 atm下为16.31 μg h-1 cm-2(-0.25 V)和5.56%(-0.2 V);30 atm下为36.87μg h-1 cm-2和12.31% (-0.2 V); 55 atm下为74.15μg h-1 cm-2和20.36% (-0.2 V)(图2d)。结果表明,随着氮气压力的增加,Rh-SA/GDY的NH3产率、FE和jNH3均显著增加。特别是在55 atm下,NH3的产率、FE和jNH3分别比室温条件下提高了7.3倍、4.9倍和9.2倍。
在室温下,同位素15N2标记实验证明,随着电解时间的延长,15NH4+信号的积分面积逐渐增大。电解6 h和12 h后产生的15NH4+产率为9.4和9.05μg h-1 cm-2;其FEs分别计算为3.35%和2.98%。这些结果与上述14N2气氛下的结果非常接近,说明电解实验中检测到的NH3确实来自NRR。最后发现高压测试体系可以适用于其他金属单原子催化剂如:Ru SA/GDY和Co SA/GDY。实验结果显示,在高压条件下,氮气还原的转换效率得到明显的提高(图2i)。
C、活性分析
接下来用DFT对Rh-SA/GDY进行了第一性原理计算,以揭示加压系统对提高ENRR活性的影响。N2在Rh-SA/GDY上有两种吸附方式:end-on吸附和side-on吸附。在50 atm的N2下,计算出前者的N2吸附自由能为-0.48 eV,后者为-0.06 eV。因此,end-on吸附的N2*比side-on吸附的N2*更稳定。而在1 atm下,N2的end-on吸附自由能计算为-0.38 eV。相比之下,50 atm下end-on的N2*形成的驱动力比1 atm下的大0.1 eV,对应于9.62 kJ/mol能量。结果表明,高压N2显著促进了N2的吸附,进而抑制了析氢反应。该工作还研究了与Rh配位的炔烃C原子的活性。在50atm下,N2在这些C位的吸附量上升了2.29 eV,比原始石墨双炔上的吸附量低1.39 eV,但比Rh位高2.77 eV。因此,Rh位点对N2的吸附和还原更为活跃。
研究了氮还原的三种可能途径,即交替、末端和酶促途径。计算的自由能表面和优化结构如图3所示。对于交替路径和末端路径,N2的吸附最终都是以-0.48 eV的吸附自由能为基础的。在0.0 V vs. RHE下,第一个氢化步骤(N2*→HNN*)上升0.86 eV。按照交替路径,HNN*→HNH*仍然吸热0.59 eV。此路径中的其他氢化步骤都是下坡(注意,NH3的解吸吸热为0.58 eV,在室温下可以超过)。因此,交替路径的电位限制步骤是限制电位为-0.86 V的第一步。
末端反应途径与交替反应途径有许多共同的中间产物,包括末端吸附的N2*和HNN*、NH2*和NH3*。沿着末端反应途径,HNN*上的末端不是HNH*而是H2NN*。此步骤仅吸热0.20 eV。随着第三个(H+ + e-)的转移,H2NN*被还原为N*,同时释放出NH3分子。该步骤需0.82 eV,略低于N2*→HNN*。N*加氢生成NH3*都是放热的。因此,远端路径的电位限制步骤仍然是第一个质子耦合电子转移步骤,其限制电位为-0.86 V。
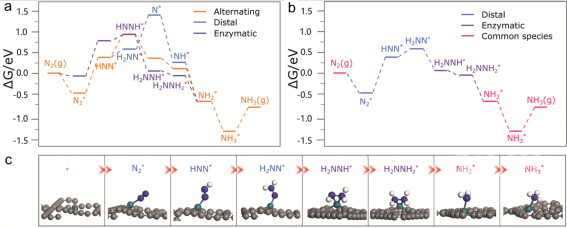
▲图3. (a)N2还原末端,交替,和酶反应途径的表面自由能。(b)低能中间体的杂化反应途径。(c)图3b中间体的优化结构。灰色、绿色、蓝色、白色球体呈现了C、Rh、N、H原子。
对于酶促途径,N2吸附模式被认为是side-on方式,吸附自由能为-0.06 eV。与交替路径类似,两个N原子也交替氢化,只是中间产物偏向吸附模式。有趣的是,N2*和HNN*更倾向end-on吸附,而H2NNH*和H2NNH2*更倾向side-on吸附端(图3b)。HNNH*的side-on的吸附能量与HNNH*的end-on的吸附能量相同,但在末端路径比H2NN*高0.39 eV。虽然N2*和HNN*上的side-on能量比在N2*和HNN*的end-on的能量高,但side-on的N2*到HNN*的还原过程上升了0.85 eV,这与交替路径的能量几乎相同。side-on的HNN*还原为HNH*的过程中有0.19 eV的吸热性。酶促途径中的其他氢化步骤都是放热的。因此,酶促径的电位限制步骤也是第一个氢化步骤,限制电位为-0.85V。
考虑到所有这些中间产物,Rh-SA/GDY很可能通过低能量的中间产物进行N2还原。例如,远端路径中的H2NN*物种可能穿梭到酶促途径,从而避免了高能中间体N*,导致side-on的H2NNH*上形成,从而降低H2NN*转化的障碍。但该杂化途径的电位限制步骤仍然是N2*→HNN*,其限制电位为-0.86 V。然而,如果酶促途径中的N2*先往返于末端途径,然后H2NN*又往返于酶促途径,则极限电位将变为-0.44 V。
总结与展望
综上所述,该工作证实了金属单原子催化剂和加压电化学测试体系的协同作用可以同时调控电催化合成氨反应的动力学和热力学过程,为电极表面传输更多的氮气分子来提高合成氨的催化活性,并降低析氢反应。所提出的策略能够协同高效的催化剂和加压反应系统,为电催化合成氨反应的实际应用提供新的思路。
来源:rationalscience 研之成理
原文链接:http://mp.weixin.qq.com/s?__biz=MzUxMDMzODg2Ng==&mid=2247546719&idx=3&sn=bab47470e4783e19307005106c69e3d5
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
原子连锁反应
单原子铜催化剂增强氧还原反应
单原子Pd1/CeO2催化剂催化芳香族碘化物烷氧基羰基化反应
原子反应

高负载量的原子级分散催化剂Ir/MoC的加氢反应性能
超高速“电子相机”捕捉处于锥形交叉点的分子

组成一切物质的原子,有一半都是来自其他星系

大型强子对撞机首次加速原子:达到接近光速

JACS封面文章:单原子催化剂应用于类芬顿反应

“催化可塑性”赋予金属铋新用途


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号