
科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2021-02-12
来源:JIPB
RNA-seq能够全面快速地获取特定样品的所有转录本的信息,并且具有基因表达水平检测范围广,技术背景噪音低的优点,目前已经成为了转录组分析的首选方案。常规的RNA-seq文库构建方法较为繁琐,通常包括mRNA的分离,mRNA片段化,反转录合成cDNA第一链,cDNA第二链合成,双链cDNA末端修复和加腺苷酸A,接头的连接,PCR富集等步骤。另外,利用常规的RNA-seq方法构建不同样品的测序文库时,每一个样品都需要独立处理。因此,针对大量样本时常规的RNA-seq方法不仅成本较高,而且费时费力。而实际中,很多的研究都需要对大量样本的转录组进行测序分析,包括绘制不同组织样本的转录组图谱,同一组织样本的转录组动态分析,以及群体中基因表达水平变异和调控分析等。
JIPB近日在线发表了中国农业大学农学院赖锦盛课题组题为“MP3RNA-seq: massively parallel 3ʹ end RNA sequencing for high-throughput gene expression profiling and genotyping”(https://onlinelibrary.wiley.com/doi/abs/10.1111/jipb.13077)的研究论文。该研究建立了一种针对大量样本的转录组文库构建和测序方法。该方法文库构建和测序的总费用约为45 RMB/样品,只有常规RNA-seq方法成本的1/10,适合于高通量的基因表达分析和基因型分析。
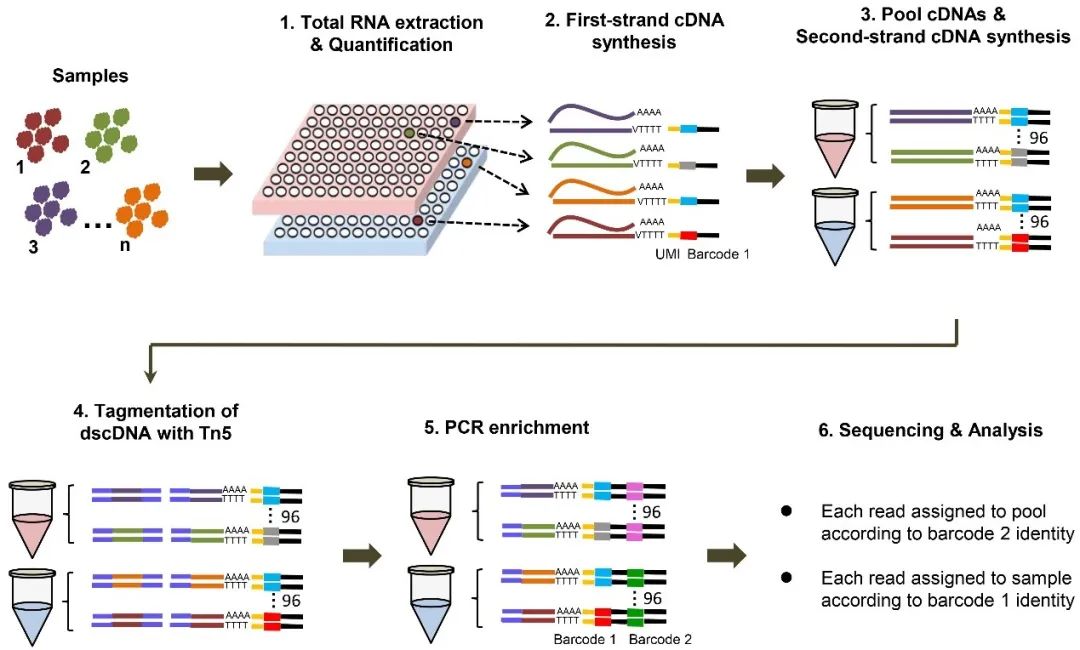
MP3RNA-seq文库构建流程示意图
赖锦盛课题组通过在反转录合成cDNA第一链的过程引入一级条形码,实现了不同待测样品文库在同一实验中的并行构建。通过在反转录合成cDNA第一链的过程引物唯一分子识别码,实现了PCR过程产生的重复序列的有效识别和去除。通过引入一级和二级条形码,以及通过基于转录本3’端对基因表达水平进行检测,实现了文库构建和测序的高通量。基于对玉米、拟南芥、人和小鼠等样品的分析表明该方法具有与常规RNA-seq方法相当的基因表达水平检测准确性和表达基因检出效率。此外,利用MP3RNA-seq方法成功的对一个包含477个DH系的玉米群体进行了基因表达水平和基因型分析,并以此为基础对基因表达调控位点和株高、穗位高和相对穗位高的调控位点进行QTL定位。研究鉴定到了25797个eQTL,其调控15335个基因。此外鉴定到了21个株高、穗位高和相对穗位高的QTL,包括一个落在2号染色体20-30 Mb的QTL,其同时影响这三个农艺性状。
中国农业大学农学院博士后陈建和博士生张湘博为该论文的第一作者,赖锦盛教授和博士后陈建为通讯作者。本研究得到了国家自然科学基金、国家重点研发计划和博士后创新人才支持计划等多个项目的支持。
原文链接:http://mp.weixin.qq.com/s?__biz=MjM5ODc5NzY4OQ==&mid=2650212402&idx=1&sn=b907e236e3407f4135b5a5a6c2978e18
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
肿瘤基因检测小知识 | 液态活检之循环肿瘤DNA
王小林/仇子龙/楼文晖团队开发基于单链游离DNA的建库和测序方法

在一天内完成样本制备到结果报告全流程的高通量基因测序平台
精彩预告 | 高通量测序技术在临床中的应用及高通量实验室的规范化建设

我科学家破解小麦遗传转化中基因型依赖难题
利用Nanopore高通量测序技术解析污水处理体系可移动抗性基因组

疾控福利~纯干货的高通量测序培训班

CSPPT研究最新分析,MTHFR基因型决定叶酸的中风预防效果
MPB:沈阳生态所李琪组-土壤线虫群落DNA提取、扩增及高通量测序
利用Nanopore高通量测序技术解析污水处理体系可移动抗性基因组


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号