科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2021-06-30
宏基因组学是直接研究环境样品中的遗传学材料,主体研究对象是微生物群落的基因组,即所有微生物个体基因组的总和。随着近20年以来DNA测序成本的大幅降低(> 105倍)和第二代高通量测序技术(NGS)大规模应用,宏基因组学(Metagenomics)正在快速发展成生物学与环境科学的一个重要的新兴交叉学科分支,其方法学正史无前例地转变着传统环境生物学与生态学的研究方式1,2。伴随着现代分子生物学方法与技术流程的不断自动化与大规模商业化,当前微生物群落研究的时间与经济成本投入已从检测通量低、劳动力密集型的分子生物学“湿实验”转变为检测通量高、数据处理密集型的计算生物学“干实验”(例如生物信息和生物统计计算)2,3。
宏基因组学是打开通往未知微生物界大门的钥匙,广义上包括环境基因组、生态基因组学和群体基因组学的研究目标与内容。自然环境中超过99%以上的微生物在已知实验室条件下尚未被分离培养。相比于传统方法(如分离与培养)与现代分子生物学技术,宏基因组学的方法与技术的最大优势体现在不依赖实验室分离培养来解码微生物组赋存的物种与基因多样性4。通过环境样品中遗传学材料的直接分离提取与测序、生物信息学、生物统计学等多方法与技术手段可全面研究微生物群落结构与功能,本文统称为“泛宏基因组”(Bulk metagenome)。不同于基于功能表型或活性筛选(简称“功能宏基因组学”)或者是仅靶向部分微生物组信息(如16S核糖体RNA基因扩增子测序、可培养微生物群落测序)的宏基因组学研究技术路线,获取环境样品的泛宏基因组信息无需对微生物进行富集或其核酸进行PCR扩增,避免了微生物可培养性、引物设计与扩增偏好性等问题的干扰,能更真实反映微生物群落在原位环境中生物多样性及组成丰度信息。
简言之,宏基因组学突破了环境微生物难培养的研究瓶颈,打开了微生物界物种和基因多样性的知识与资源宝库,促进了包括新物种、新酶基因(如CRISPR-Cas核酸酶)、新功能代谢过程(如完全氨氧化)与新活性物质(如抗生素)的发现。此外,针对自然或工程生态系统的大规模时间与空间尺度宏基因组学研究有助于探明微生物之间及微生物与环境之间的复杂相互作用,进而阐释群落构建机制,完善群落生态学理论4-8。过去10余年基于高通量测序的宏基因组学方法与工具已被广泛应用于生物学、环境科学等领域的前沿交叉学科研究,衍生了一系列新发现与重大成果;其中,代表性的研究成果可大致归纳为以下五类:(1)新微生物物种的发现;(2)新酶基因(簇)及功能的发现;(3)新物质代谢途径的发现;(4)微生物组-环境-效能新关联的发现;(5)新生命进化树与物种分类新规则。
通过不同类型环境样品采集、核酸提取、宏基因组测序、基因组重构等多技术手段联用有助于不断发现新的微生物物种。美国加州大学伯克利分校Jill Banfield教授是环境宏基因组学领域的先驱者与引领者9,10。她的团队通过利用宏基因组组装基因组(Metagenome-Assembled Genome, 简称MAG)分析与转录组分析研究了科罗拉多河附近蓄水层的微生物组,发现了一类 “不寻常微生物”,命名为“候选门辐射群” (Candidate Phyla Radiation,简称CPR) 11 。这类CPR微生物不同于传统观念中的原核生物:它们的基因组编码了外显子与内含子,还具有自我剪接的内含子及在其核糖体(rRNA)蛋白质编码基因。该发现更新了领域对原核生物组成的理解,相关成果于2015年在《自然》上发表后引发了国际同行对包括CPR在内的未培养微生物暗物质的进一步关注和后续研究。
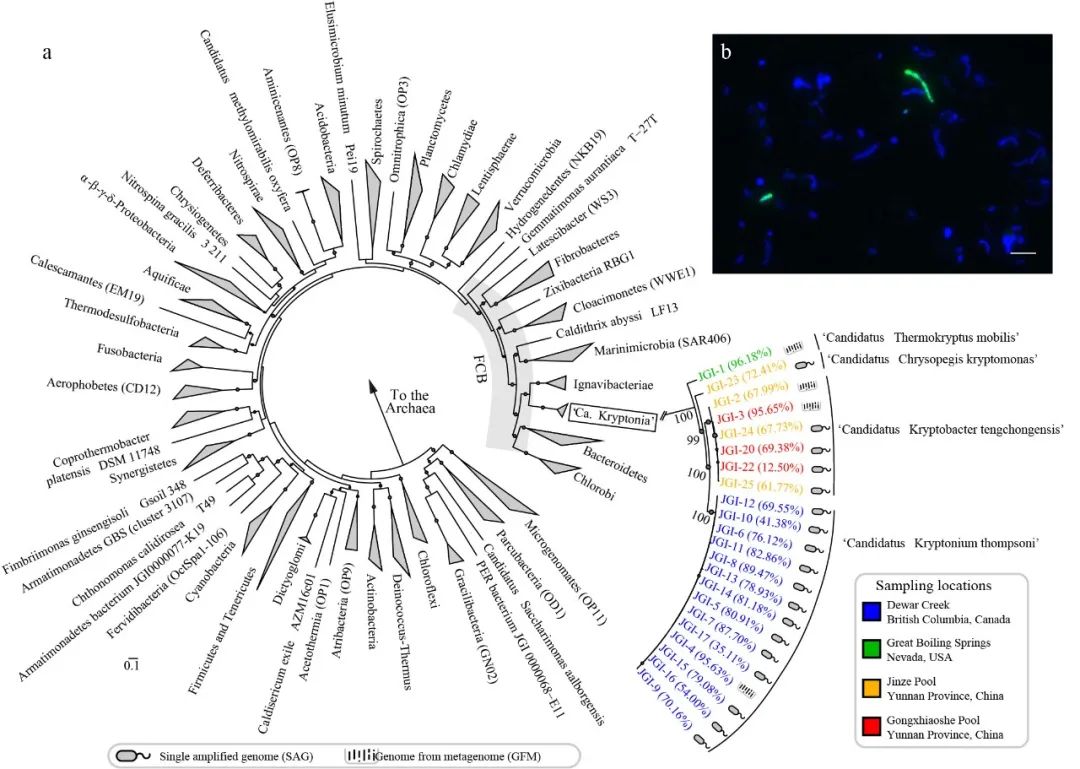
图1. 候选门Kryptonia的发现。(a) 基因组水平关联物种的系统发育树;(b)Candidatus Kryptonia在热泉沉积物中的荧光原位杂交(绿色)影像12。图片设计取材于文献。
宏基因组测序研究产生了海量可开源获取的环境微生物组和动物微生物组序列。相应地,针对全球主流公共数据库中宏基因组大数据的宏分析(Meta-analysis)研究稳步成为新知识与新发现的重要途径。例如:来自美国能源部联合基因研究所JGI的研究团队通过JGI-IMG/M 数据库中5.2 TB宏基因组数据的重分析与深度挖掘12,于2016率先发现了一种新的候选细菌门Candidatus Kryptonia(图1)。研究结果表明这类未培养微生物为异养菌,主要在高温、 pH呈中性的地热泉中存在。该项研究是通过全球公共数据库中可开源获取宏基因组数据重分析与深度挖掘来发现新的微生物物种及其基因组编码的功能潜能的典型范例。
通过环境样品的大规模宏基因组学研究还有助于发现新的酶基因系统以及生物合成基因簇。美国UC Berkeley 地球与环境科学系Jill Banfield教授和生物化学与分子生物学系Jennifer Doudna教授的团队开展交叉学科合作,解析了来自地下水、底泥、生物膜、土壤以及婴儿肠道等环境微生物样品中TB级别的宏基因组序列;通过组装、分箱等深度挖掘手段从得到了1.55亿个蛋白编码基因和大量MAGs序列13。该研究通过未培养微生物基因组信息的深度挖掘,发现了第一个来自古菌域的Cas9蛋白,还在未培养细菌中发现了两种新的CRISPR-Cas系统,并将其命名为CRISPR-CasX和CRISPR-CasY系统,该成果于2017年发表在《自然》13。Doudna教授及其合作者率先在基于CRISPR-Cas系统的基因编辑领域取得了重大原创性突破,被授予2020年授予诺贝尔化学奖。目前,CRISPR-Cas9已发展成真核生物基因编辑“神器”。宏基因组学为将来从未培养环境微生物中发现了更多新CRISPR-Cas系统提供了强有力的工具,进一步拓宽了识别DNA序列的范围,为发展CRISPR-Cas系统以更好应用于生物技术领域和疾病治疗等提供了更多的选择与可能。
新功能基因簇的发现还推动了新型抗菌剂的研发,当今大多数已知的细菌天然产物(包括抗生素)是从能被成功分离培养的放线菌、变形杆菌、 芽孢杆菌分离物中获取的,但它们仅占自然界中微生物群落的极少数,还有更多可以合成新抗生素的功能基因簇等待被发现和使用。例如,研究者通过大规模重建土壤中未培养微生物的基因组草图,发现了大量可合成如抗生素、抗真菌剂、免疫抑制剂等化合物的新型生物合成基因簇。来自美国洛克菲勒大学的研究团队利用扩增子测序结合BAC 文库筛选的方法从 2000 个土壤样本中发现了一个未知的抗生素合成基因簇并利用该基因簇合成了一种可杀灭多重耐药的革兰氏阳性致病菌的新型抗生素,将其命名为“马拉西啶”(Malacidins)并进一步对其结构功能和作用机制进行了深入研究(图2)。因此,通过基于宏基因组学研究可以进一步拓展对于自然界中微生物生产抗菌类次级代谢产物合成能力的认知,进一步结合功能基因组学方法验证合成基因簇的功能活性为新型抗生素的发现提供了途径。
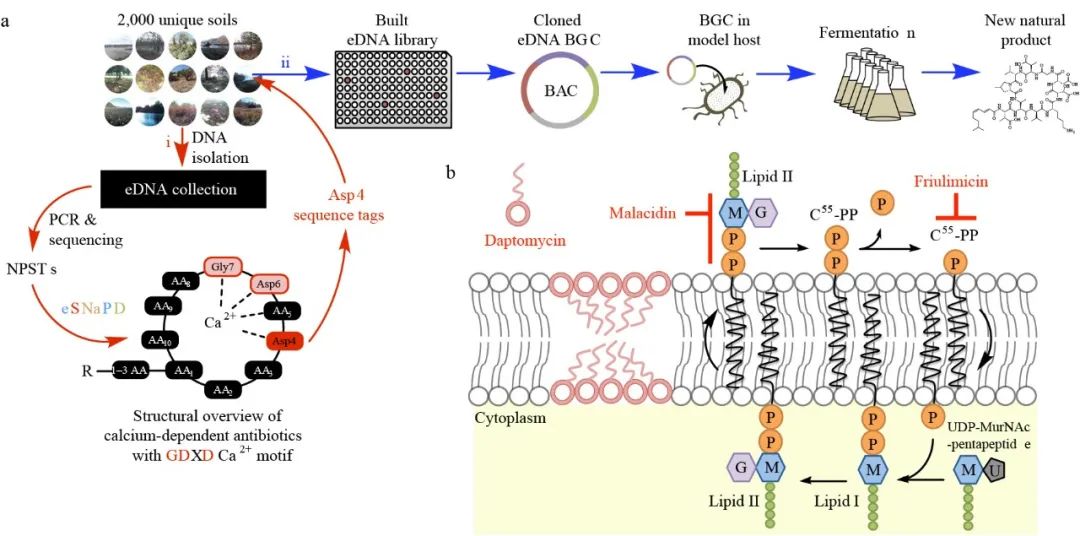
图2. 新型抗生素Malacidins的发现。 (a) 基于扩增子测序和克隆文库筛选从土壤微生物群落中筛选并发现新型钙依赖环肽抗生素Malacidin的过程 (b)新型钙依赖环肽抗生素Malacidin的作用机制 14。图片设计取材于文献。
除了有助于新功能基因与酶系统的发现,宏基因组学还推动了领域对于未培养微生物的物质与能量代谢以及在全球生物地球化学循环中作用的全新认知。比如,2015年奥地利Wagner Michael教授和荷兰Sebastian Lücker教授分别领导的独立研究团队《自然》杂志同期发表论文,声称发现了完全氨氧化(Comammox)微生物的存在 15,16。这两项独立研究均基于实验室反应器分别富集了来自水产养殖系统生物过滤器的生物膜和被热水覆盖金属管道表面的微生物生物膜样品,再基于宏基因组测序、同位素示踪技术结合MAG分析,最终证实了完全氨氧化Nitrospira具备独立将氨氮氧化为硝酸盐的能力。这一新发现解开了困扰微生物学界100多年的谜题,打破了“硝化作用过程需要氨氧化菌与亚硝酸盐氧化菌两种不同微生物分两步完成”的传统观念(图3),为领域探索Comammox在氮元素的生物地球化学循环过程中的作用奠定了基础。此外,基于宏基因组测序和MAG分析的方法也推动了硝酸亚还原厌氧甲烷化过程 、深古菌门等未培养微生物参与甲烷代谢过程的发现 17,18。简言之,将宏基因组学技术与生物反应器富集、微生物原位表征技术(例如稳定同位素示踪)等有机结合,是发现新功能微生物和揭示其代谢途径和生态功能的强有力手段与重要途径,可有效推动对未培养微生物的功能与代谢潜力的认知。
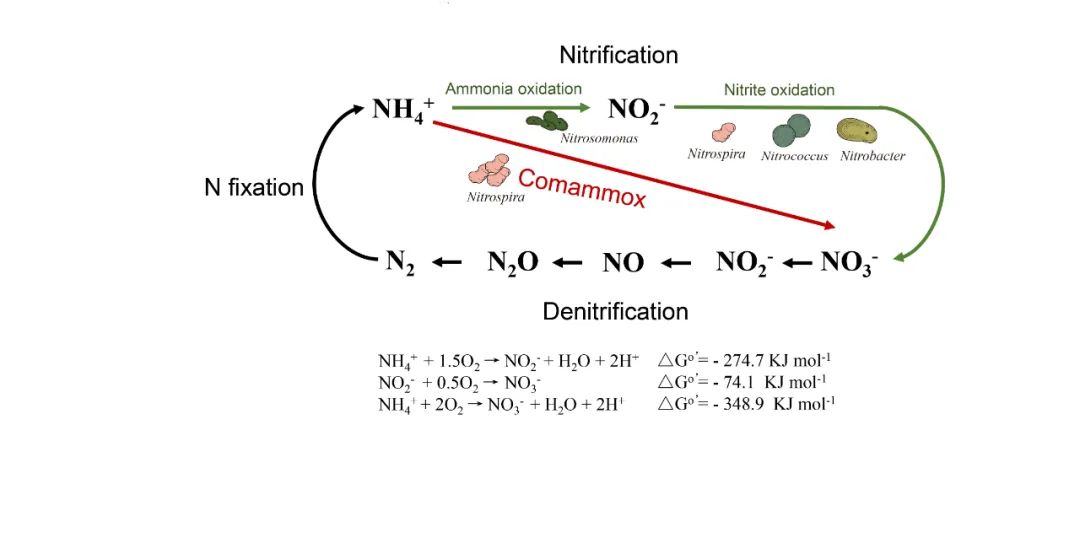
图3. 完全氨氧化(Comammox)微生物独立完成硝化过程两步反应
宏基因组学揭开了自然界中微生物群落组成和功能多样性面纱,有望推动微生物组科学向生物工程技术应用的转化。例如通过土壤微生物群落组成定向提高作物的生产力19,通过刺激自然产生或者人工投加微生物菌剂修复受污染的水体20;通过富集培养特定功能的微生物实现从废水中回收资源21等过程。探索微生物组、地球环境、人类系统之间内在固有关联,有助于进一步将关于微生物群落结构与功能的微生物组科学知识更好的应用于服务人类社会经济发展,以提高人类生活水平。例如,2015年来自香港大学研究人员基于细菌16S 核糖体RNA扩增子测序,结合共现网络模型分析、多参数统计学分析等手段解析了城镇污水处理厂活性污泥工艺中的微生物组(M)、环境参数(E;如污水水质、运行参数等)以及系统效能(P)三者之间的内在关联,并基于此提出了通过微生物组调控来减轻活性污泥起泡问题的工程措施与系统设计策略 6。近期,来自美国长期从事工程微生物组研究的科研团队提出了基于“设计-建造-测试-学习(DBTL)”的研究和技术开发体系,为微生物组在医药、农业、能源和环境方面的广泛应用提供了理论指导 22。其中,宏基因组学技术在捕获必要的生态学原理,把控微生物组群落组成,预测的群落功能等相关的设计、建造和测试环节中都发挥了重要作用。
真核生物的起源是进化生物学界研究的谜题与热点23。近年来瑞典乌普萨拉大学Thijs Ettema教授实验室24,25和美国加州大学伯克利分校 Jill Banfield教授实验室10,26基于环境样品采集、宏基因组学测序、大规模基因组构建与比较基因组学宏分析,发现了大量未培养环境微生物类群,并基于此提出了真核生物起源于古菌的新证据(图4)。例如:2015年,Spang等在《自然》发表文章,他们采集了北冰洋大洋中脊受到热液作用的深海沉积物样品,对其进行16S 核糖体RNA基因序列的宏基因组学分析,发现了一个全新的微生物门类名为Lokiarchaeota,系统发育树分析表明这是当时已知与真核生物亲缘关系最近的古菌生物,该发现填补了原核生物向真核生物过渡的进化学研究空白24。在后续的研究中,Odinarchaeota、Heimdallarchaeota和 Thorarchaeota也陆续被发现,它们与真核生物一同构成了Asgard 古菌门,并被认为是真核生物起源的谱系25。结合之前对TACK古菌的研究,新的真核生物起源的系统发生树被勾勒出来。这些发现表明真核生物起源于古菌,改写了从1977 年以来学界普遍认为的生命体划分为“真核生物、细菌和古菌”三大界的历史27。与此同时,Jill Banfield教授课题组也通过MAG 分析和全基因组的完整核糖体蛋白的系统发育分析提出生命系统可以重新划分为 2 个域(图4),这个新的系统发育关系也表明真核生物可以很好的从古细菌内部演化而来24,26。
近年来,全球公共数据库中储存的宏基因组数据量在不断扩大,促使领域学者可以更好的利用来自不同环境的微生物样品的基因组序列开展宏分析研究,并基于此提出关于生命进化树的新见解,开发新工具。微生物的生命进化树与物种分类信息相互关联。昆士兰大学Hugenholtz 教授课题组通过使用公认的平均核苷酸相似度(Average Nucleotide Identity, ANI)标准来设置物种界限并提出包含所有公共可获得的细菌和古细菌基因组的物种簇, 提出了一个可以为细菌和古细菌基因组提供完整的从域到种的分类学新框架。在这个新的分类系统下, NCBI 数据库中 58% 的物种分类信息将被更改,这也预示着将会有更多的对物种基因水平界限和相关生态学以及进化研究的新认知产生 28。目前,宏基因组数据分析中常用于基因组分类的GTDB数据库是基于单系和亲缘系进化的差异加以限定的细菌和古细菌类群基因组的广泛分类法,其中大约40%的基因组在NCBI数据库中缺乏种名 29。
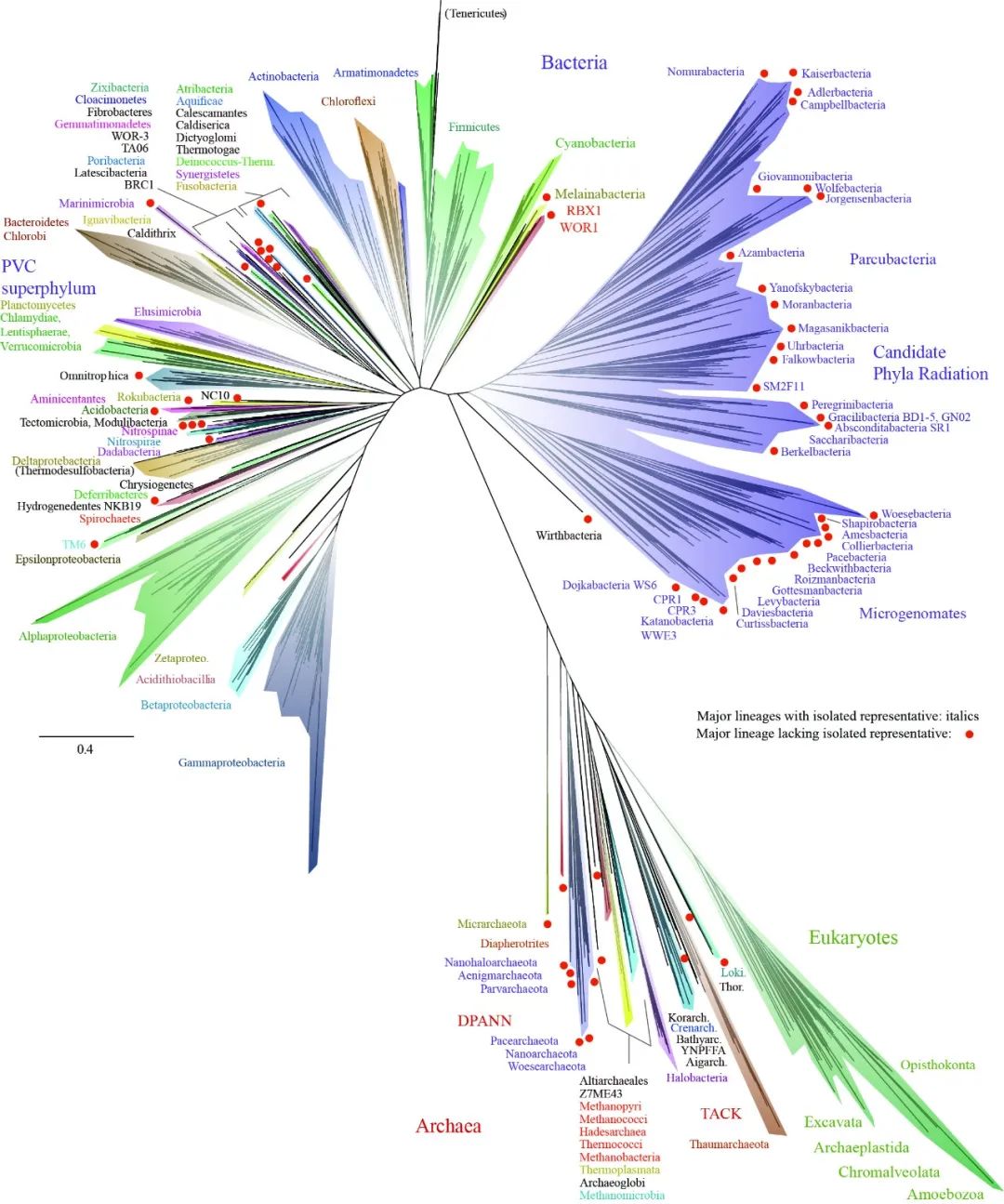
图4. Banfield课题组构建的2个域系统发育关系树。图片设计取材于文献24,26
尽管近10年宏基因组学在生物学与环境生态学研究中的应用催生了一系列重大的新发现,但是我们对环境赋存海量未培养微生物的认识依旧有限,仍然需要进行不断的方法学改进与理论探究。显而易见,微生物样品的收集、DNA提取以及宏基因组测序等过程会对组学研究结果产生影响,因此在制定标准规范化的操作流程的同时,针对不同特点的微生物样品的DNA提取等分子生物学流程进行优化以避免研究结果的偏差是必要的1。目前领域学者可较为容易的通过宏基因组组装重构大量基因组草图(MAGs)。尽管通过未培养微生物基因组学分析可有效预测其功能与代谢潜能以及微生物群落内物种相互作用,但是DNA水平的潜能不等同于功能与活性。因此,迫切需要新的微生物组学研究手段实现微生物组成与功能的耦合研究。鉴于此,需要进行更多的研究以提供原位生理功能测定,将微生物群落的物种与基因多样性信息与真实功能进行关联。目前已有多种方法可以用来量化微生物群落的活性,例如稳定性同位素探针技术30、转录组学31、培养组学32、代谢组学33和蛋白质组学34。随着更多的表征手段和新方法的有机结合,宏基因组学必将为环境微生物学领域研究带来更多的新发现和新惊喜。
参考文献:
1 Ju, F. & Zhang, T. Experimental design and bioinformatics analysis for the application of metagenomics in environmental sciences and biotechnology. Environ. Sci. Technol. 49, 12628-12640 (2015).
2 Liu, Y.-X. et al. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell (2020).
3 Ju, F. & Zhang, T. 16S rRNA gene high-throughput sequencing data mining of microbial diversity and interactions. Appl. Microbiol. Biotechnol. 99, 4119-4129 (2015).
4 刘永鑫, 秦媛, 郭晓璇 & 白洋. 微生物组数据分析方法与应用. 遗传 41, 845-862 (2019).
5 鞠峰 & 张彤. 活性污泥微生物群落宏组学研究进展. 微生物学通报 46, 2038-2052 (2019).
6 Ju, F. & Zhang, T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. The ISME journal 9, 683 (2015).
7 Ju, F., Xia, Y., Guo, F., Wang, Z. & Zhang, T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ. Microbiol. 16, 2421-2432 (2014).
8 Hu, A. et al. Strong impact of anthropogenic contamination on the co‐occurrence patterns of a riverine microbial community. Environ. Microbiol. 19, 4993-5009 (2017).
9 Tyson, G. W. et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37-43 (2004).
10 Castelle, C. J. & Banfield, J. F. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172, 1181-1197 (2018).
11 Brown, C. T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208-211 (2015).
12 Eloe-Fadrosh, E. A. et al. Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat. Commun. 7, 1-10 (2016).
13 Burstein, D. et al. New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237-241 (2017).
14 Hover, B. M. et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nature microbiology 3, 415-422 (2018).
15 Daims, H. et al. Complete nitrification by Nitrospira bacteria. Nature 528, 504-509, doi:10.1038/nature16461 (2015).
16 van Kessel, M. A. et al. Complete nitrification by a single microorganism. Nature 528, 555-559, doi:10.1038/nature16459 (2015).
17 Haroon, M. F. et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500, 567-570, doi:10.1038/nature12375 (2013).
18 Evans, P. N. et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434, doi:10.1126/science.aac7745 (2015).
19 de Vries, F. T., Griffiths, R. I., Knight, C. G., Nicolitch, O. & Williams, A. Harnessing rhizosphere microbiomes for drought-resilient crop production. Science 368, 270, doi:10.1126/science.aaz5192 (2020).
20 Pi, K. et al. Remediation of arsenic-contaminated groundwater by in-situ stimulating biogenic precipitation of iron sulfides. Water Res 109, 337-346, doi:10.1016/j.watres.2016.10.056 (2017).
21 Yuan, Z., Pratt, S. & Batstone, D. J. Phosphorus recovery from wastewater through microbial processes. Curr Opin Biotechnol 23, 878-883, doi:10.1016/j.copbio.2012.08.001 (2012).
22 Lawson, C. E. et al. Common principles and best practices for engineering microbiomes. Nat Rev Microbiol 17, 725-741, doi:10.1038/s41579-019-0255-9 (2019).
23 Williams, T. A., Foster, P. G., Cox, C. J. & Embley, T. M. An archaeal origin of eukaryotes supports only two primary domains of life. Nature 504, 231-236 (2013).
24 Spang, A. et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173-179 (2015).
25 Zaremba-Niedzwiedzka, K. et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541, 353-358 (2017).
26 Hug, L. A. et al. A new view of the tree of life. Nature microbiology 1, 1-6 (2016).
27 Eme, L., Spang, A., Lombard, J., Stairs, C. W. & Ettema, T. J. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 15, 711 (2017).
28 Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36, 996-1004, doi:10.1038/nbt.4229 (2018).
29 Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol 38, 1079-1086, doi:10.1038/s41587-020-0501-8 (2020).
30 Mooshammer, M. et al. Flow-through stable isotope probing (Flow-SIP) minimizes cross-feeding in complex microbial communities. ISME J, doi:10.1038/s41396-020-00761-5 (2020).
31 Oyserman, B. O., Noguera, D. R., del Rio, T. G., Tringe, S. G. & McMahon, K. D. Metatranscriptomic insights on gene expression and regulatory controls in Candidatus Accumulibacter phosphatis. ISME J 10, 810-822, doi:10.1038/ismej.2015.155 (2016).
32 Wang, Q. & He, J. Complete nitrogen removal via simultaneous nitrification and denitrification by a novel phosphate accumulating Thauera sp. strain SND5. Water Res 185, 116300, doi:10.1016/j.watres.2020.116300 (2020).
33 Qiu, G. et al. Metabolic Traits of Candidatus Accumulibacter clade IIF Strain SCELSE-1 Using Amino Acids As Carbon Sources for Enhanced Biological Phosphorus Removal. Environ Sci Technol 54, 2448-2458, doi:10.1021/acs.est.9b02901 (2020).
34 Sukul, P. et al. Simple discovery of bacterial biocatalysts from environmental samples through functional metaproteomics. Microbiome 5, 28, doi:10.1186/s40168-017-0247-9 (2017). 来源:宏基因组
原文链接:http://mp.weixin.qq.com/s?__biz=MzUzMjA4Njc1MA==&mid=2247500684&idx=1&sn=7abad3acf0abf9549cbad25fc8d5ed27
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
“垃圾DNA”对于染色体组连接至关重要

中国人类基因组研究有望“弯道超车”
我国学者在合成型基因组重排领域取得重大突破



小麦D基因组完整图谱首绘成功

西安交大在《科学》发文揭示鸦片罂粟基因组及吗啡合成原理

中国研究团队破解中国种茶树全基因组密码

迄今最大合成基因组诞生:非全部密码子构建

质体基因组工程新策略:微型合成质体的开发

华中农业大学教授赵书红:在基因组上“跳舞”
利用Nanopore高通量测序技术解析污水处理体系可移动抗性基因组


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号