科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-01-16
来源:BioArt
撰文 | 奚望
责编 | 兮
蛋白质结构预测一直是热门的基本问题,因为结构决定了功能。传统的蛋白结构预测主要依赖片段组装法(fragment assembly)。人们通过分析PDB(Protein Data Bank)等数据库中的蛋白结构得到某种统计势能,然后以此为目标,通过不断地随机采样来选择最小化该目标势能值的结构。片段组装法在实施中通常是每一步改变结构的一小部分,而保留其余的大部分,因此需要经过上千步和多次重复才能获得理想结构。
谷歌公司深度学习方面的核心团队DeepMind(是一家英国的人工智能公司。公司创建于2010年,最初名称是DeepMind科技——DeepMind Technologies Limited,在2014年被谷歌收购),一直致力于用人工智能和神经网络技术解决不同场景下的学习问题。继围棋博弈算法AlphaGo之后,DeepMind转向了基于氨基酸序列的蛋白质结构预测,提出了名为AlphaFold的深度学习算法,并在国际蛋白质结构预测比赛CASP13(Critical Assessment of ProteinStructure Prediction)中取得了优异的成绩。
2020年1月15日,DeepMind团队Andrew W. Senior等在Nature发表文章 Improvedprotein structure prediction using potentials from deep learning,展示了AlphaFold算法的细节和表现。

近年来蛋白结构预测领域出现了一种基于DNA序列协同进化(evolutionary covariation)的新方法。人们先找到和目标序列相似的DNA序列,并进行多序列比对(multiple sequence alignment,MSA),其中序列变化存在相关性的位点就存在相互接触的可能。借助一些统计方法,比如神经网络,就可以从序列比对的数据中预测出氨基酸残基的接触概率。
AlphaFold算法流程如图所示:以目标序列本身和目标序列与相似序列的比对结果为特征输入神经网络,首先通过卷积神经网络进行特征提取,学习到这些蛋白的势能分布。接着,只要通过梯度下降法,就能获得对于目标序列本身势能最低的结构。预测的结果包括蛋白骨架的扭转角度和两两残基之间距离。
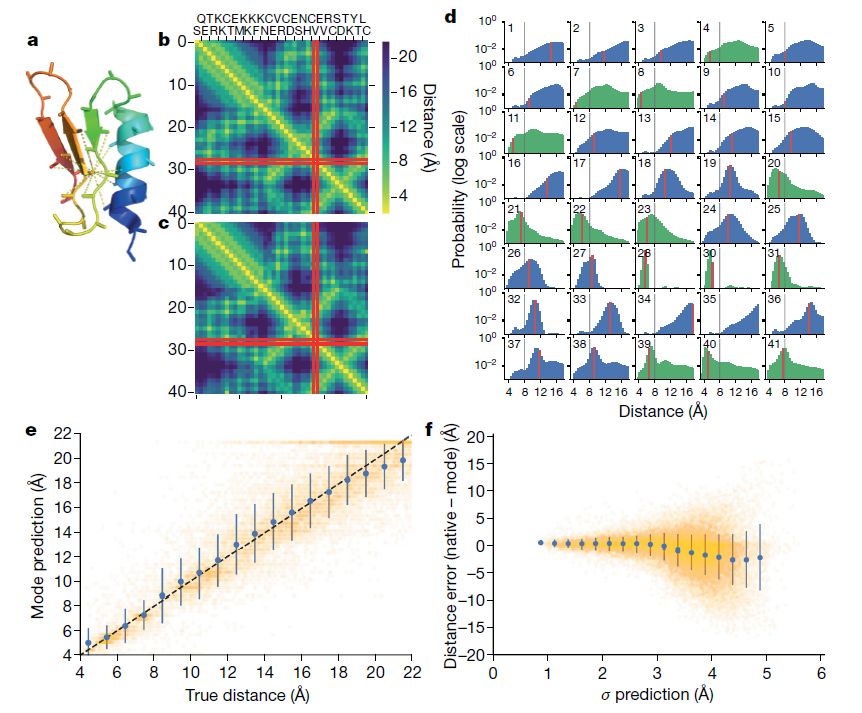
卷积神经网络部分大小为64x64,即每一步推断64个残基间的相互距离。以CASP上的T0955为例,通过PDB数据库上所有蛋白结构的训练后,神经网络作出了和实际残基距离分布非常相似的精确预测。该模型同时也能输出预测的不确定性大小。
在获得残基间距离后,作者据此构建了一个势能Vdistance。他们将该势能表达为骨架转角的函数,并使用L-BFGS梯度下降法逐步逼近势能最小值,并从而获得相应的骨架转角度数。预测的精确度(TM分值)随着梯度下降的步数迅速增加。
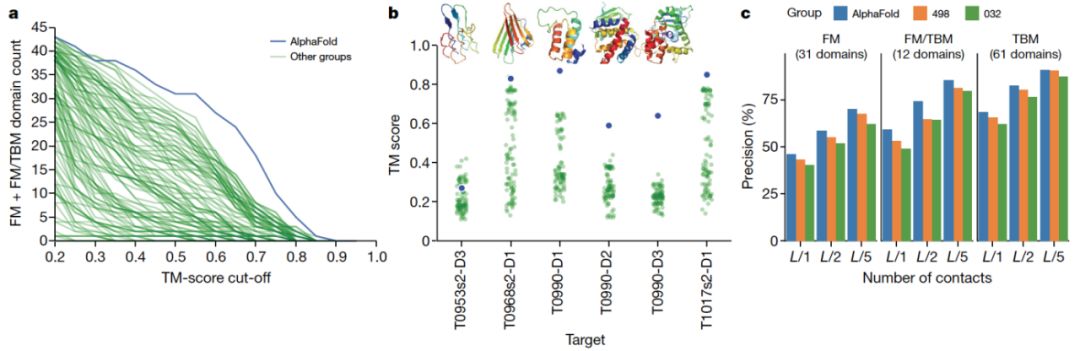
在CASP比赛中,相较于其他算法,AlphaFold在同样条件下能显著地预测出更多不存在同源结构的新结构。而无论同源结构是否存在,AlphaFold预测精确度也都比另外两个之前的最优模型Raptor-X Contact和TripletRes要高。
作者据此认为,AlphaFold的成功显示了一个经过仔细设计的深度学习模型是可以同时完成预测蛋白质残基间距离和蛋白骨架转角的任务。进一步发展该类模型有利于推进蛋白结构预测的进步。
原文链接:
https://doi.org/10.1038/s41586-019-1923-7
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652479973&idx=4&sn=7452b237c29bd6c0796b22dba24abddd&chksm=84e23251b395bb475be3bbab6d5877d69887be996251eb845a99780212d60a0e09d1a9a7bd83&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
曾坚阳团队开发蛋白-小分子相互作用预测的深度学习模型
可视化深度学习揭示基因如何影响细胞特征
深度学习框架
“阿尔法折叠2”预测蛋白结构近原子水平

借助机器学习,对垃圾信息说“不”

深度学习方法可鉴定涉及组织再生和癌症的新基因

图灵奖后,深度学习呼唤深度理解

人工智能代替人类?还有很长的路要走

AI芯片设计难?厂商要给软件开发者降低门槛
深度学习仿真工具


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号