来源:X一MOL资讯
如何高效地获得手性分子的两种对映异构体一直是有机合成、药物化学、材料科学等领域的基本挑战之一。在过去的几十年里,不对称合成得到了巨大的发展,已成为一种制备光学纯化合物的强大工具。原则上,通过切换手性组分(如手性底物、辅助剂、试剂、催化剂和配体)的构型,可以合成目标分子的不同对映异构体(图1a)。但是,如果这些手性组分源于自然界(仅以单一对映异构体形式存在),那么情况可能会变得复杂。另一方面,反应时长长短也是不对称合成中的一个重要变量。动力学拆分(KR)是利用一对对映异构体与手性试剂或催化剂的反应性差异,得到光学纯的目标产物和未反应底物的混合物,转化率约为50%。在更复杂的平行动力学拆分(PKR)中,两个KR反应同时进行,因此可以通过两个平行的反应路径从外消旋底物得到不同的光学纯产物的混合物。尽管反应时长会影响目标分子的光学纯度,但产物的绝对构型仍由手性试剂或催化剂决定。

图1. 手性分子相反构型对映体的合成。图片来源:Nat. Chem.近日,中国科学院上海有机化学研究所的游书力研究员课题组报道了一种反应时长依赖的对映发散性合成(time-dependent enantiodivergent synthesis)方法,即在相同的手性催化体系下,通过控制反应时长,便可分别得到目标产物的一对对映异构体(图1b)。具体而言,一种手性催化剂可以促进两个KR反应顺序进行。当在不同的特定反应时间点淬灭反应体系时,便能分别分离出高对映体纯度的相反构型手性产物。相关成果发表在Nature Chemistry 上,第一作者为涂航飞。
作者在进行6-羟基异喹啉(1a,1.0当量,0.2 mmol)和叔丁基(1-苯基烯丙基)碳酸酯[(rac)-2a,2.0当量]的不对称分子间烯丙基胺化反应时(图2a),发现当使用[Ir(cod)Cl]
2(3 mol%)和Carreira手性亚磷酰胺配体(S)-L1(12 mol%)衍生的Ir催化剂时,并向反应中加入3,5-二氯苯甲酸(30 mol%)作为添加剂,在室温下于MeOH(0.1 M)中平稳进行10 h后,胺化产物(R)-3aa的收率为78%,e.e.值为98%(条件A)。有意思的是,当反应在没有Brønsted酸添加剂的情况下进行到6 min时淬灭,能以80%的收率和94%e.e.值获得对映体(S)-3aa(条件B)。
为了探究反应机理,作者进行了一系列机理研究。首先,将(S)-3aa(93%e.e.)与2.0当量的(rac)-2a反应10h,以75%的收率和88%的e.e.值得到(R)-3aa,以及以53%的收率和90%的e.e.值得到肉桂基甲基醚(S)-4a(图2b)。由于烯丙基胺可通过醇溶剂中的氢键活化而参与过渡金属催化的烯丙基取代反应,因此作者推测2a和3aa均存在KR反应(图2c、2d)。为了证实该假设,作者将(rac)-2a、(S)-2a和(R)-2a分别置于条件A下,(S)-2a和(R)-2a均表现出显著变化的反应性。当(rac)-2a的反应在6 min内淬灭时,以41%的收率、93%的e.e.值得到(S)-3aa,而(R)-2a以43%的收率回收,e.e.值为97%。此外,从(S)-2a(> 99%e.e.)到(S)-3aa(82%收率和95%e.e.),以及从(R)-2a(99%e.e.)到(R)-3aa(74%的收率和94%的e.e.)均观察到手性保持,但是后者的速度要慢得多(6 min vs 10 h)。当将(rac)-3aa在条件A下搅拌8 h时,以38%的收率和93%e.e.得到(S)-4a,而(R)-3aa以54%的收率和82%e.e.回收。化合物(R)-3aa在(S)-L1存在下反应要慢得多,但当与衍生自(R)-L1的催化剂反应时,可以完全转化为(R)-4a(99%收率和99%e.e.),并以高收率(93%)分离出释放的6-羟基异喹啉1a。
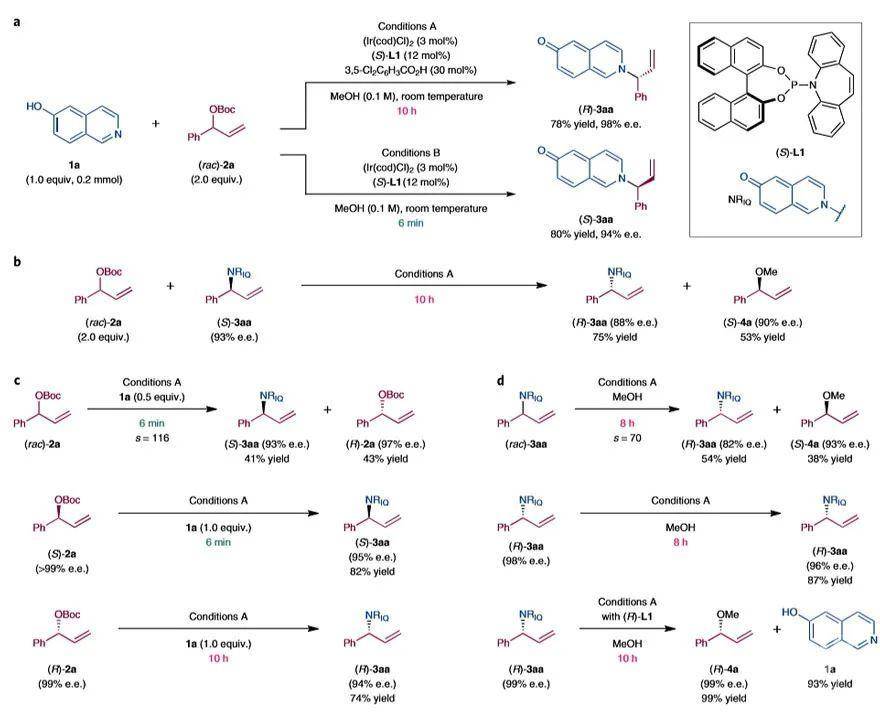
图2. 反应时长依赖的对映发散性合成的发现。图片来源:Nat. Chem.此外,作者还通过
19F NMR测量了氟取代化合物(S/R)-2b和(S/R)-3ab的反应初始速率(r)(图3a)。结果进一步证实了双重KR反应过程。即使在低温(0 °C)下,(S)-3ab的生成最快,具有最大的初始速率(r
1S = 9.89×10
-4 mol l
-1 min
-1)。而(S)-3ab的初始消耗速率和(R)-3ab在室温下的生成速率较小,但数量级相同(r
2S = 9.58×10
-4 mol l
-1 min
-1和r
1R = 2.76×10
-4 mol l
-1 min
-1)。室温下(R)-3ab的消耗要慢得多,初始速率要小得多(r
2R = 9.46×10
-6 mol l
-1 min
-1)。总的来说,这些结果清楚地表明,两个连续KR反应的存在使得以反应时长为关键参数来制备烯丙基胺的两种对映体成为可能。
如图3b所示,作者提出了可能的反应机理。以3aa为例,最初,(rac)-2a的KR通过Ir催化的不对称烯丙基胺化反应进行。由于(S)-2a的活性较高,可以在6 min内与6-羟基异喹啉反应生成(S)-3aa,而(R)-2a在此时段内(k
1S≫ k
1R)很大程度上保持不变。然而,随着反应的进行,(S)-3aa在该催化体系下会与MeOH反应,生成(S)-4a。同时,反应活性较低的(R)-2a与6-羟基异喹啉反应,逐渐积累(R)-3aa。由于(R)-3aa很稳定,不易与MeOH发生反应(k
1R≈ k
2S >>k
2R),因此可以在更长的反应时长(10 h)内以高光学纯度获得(R)-3aa。
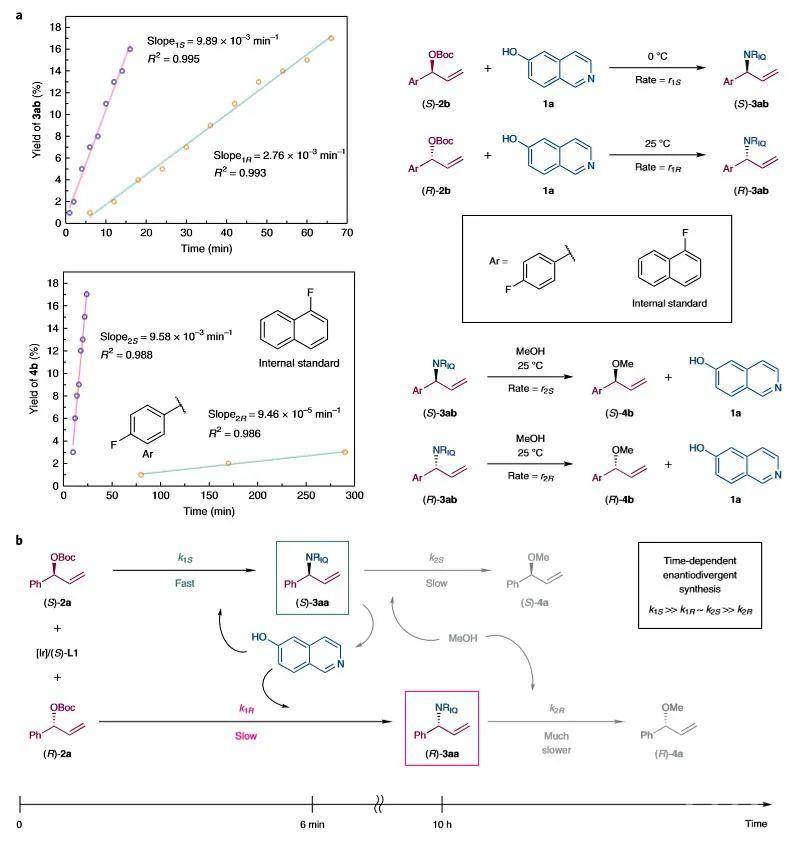
图3. 反应时长依赖的对映发散性合成的机理。图片来源:Nat. Chem.接着,作者对烯丙基胺3的对映发散性合成的底物范围进行了考察(图4)。对位、间位或邻位带有卤素、甲基和甲氧基的支链碳酸肉桂酯都能与6-羟基异喹啉反应。在条件(A)(t
1= 9-11 h)或B(t
2 = 6-10 min)下,胺化产物3aa-3ai的(R)-和(S)-对映体均具有良好的收率((R)-对映体:54-78%,(S)-对映体:51-80%)和对映体纯度((R)-对映体e.e.值:97–99%,(S)-对映体e.e.值:88–94%)。此外,该反应还能耐受2-萘基、4-联苯和3-噻吩,以较好的收率和对映选择性((R)-对映体,t
1= 10 h,收率:61-71%,e.e.值:98%;(S)-对映体,t
2 = 5-10 min,收率:56-73%,e.e.值:86-94%)得到相应的产物3aj-3al。具有苯胺基的烯丙基碳酸酯也能转化为所需的产物(R)-3am(77%的收率和99%e.e.,t
1 = 11 h)和(S)-3am(68%的收率和85%e.e.,t
2 = 6 min)。不过,碳酸巴豆酯的反应活性在胺化和醚化反应中均减弱。当反应30 min(45%的收率和92%e.e.)或18 h(76%的收率和74%e.e.。)时,仅获得(R)-3an。
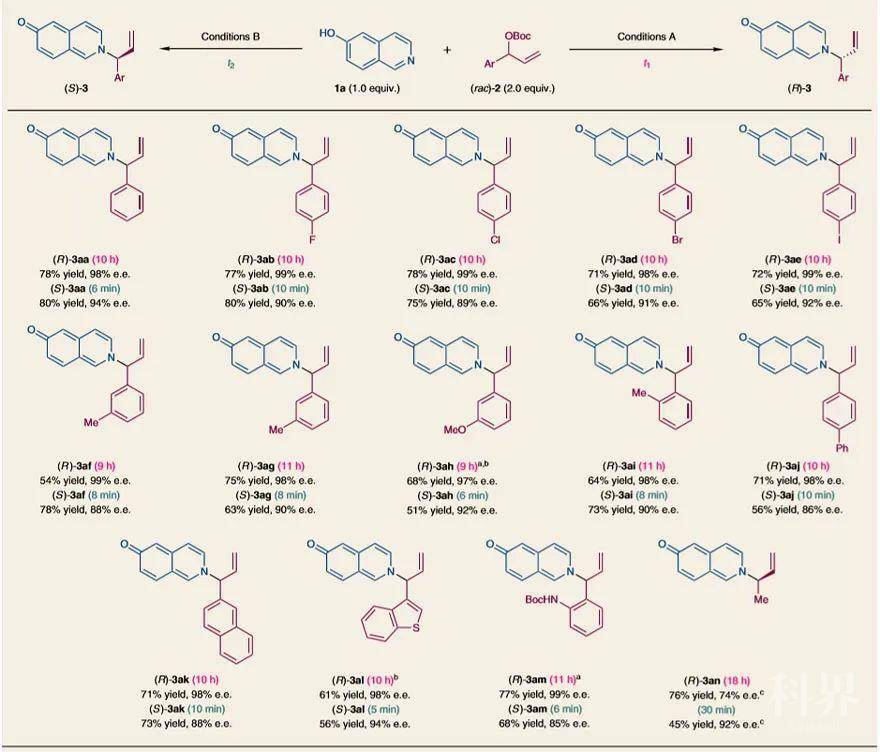
图4. 烯丙基碳酸酯底物范围。图片来源:Nat. Chem.接下来,作者考察了羟基异喹啉的底物范围(图5)。在C7或C8位具有甲基或卤素原子的6-羟基异喹啉能平稳地转化为3ba-3fa的两种对映异构体((R)-对映体,t
1= 9-11 h,收率:55-83%,e.e.值:95-> 99%;(S)-对映体,t
2 = 8 min,收率:66-82%,e.e.值:90-94%)。值得一提的是,8-羟基异喹啉也能兼容该反应,以较好的收率和对映选择性((R)-对映体,t
1 = 8-10 h,收率:77-78%,e.e.值:91-92%;(S)-对映体,t
2 = 6 min,收率:74-87%,e.e.值:91%)得到目标产物3ga和3ha。然而,5-羟基喹啉在该条件下则不能反应。此外,反应时长依赖的对映发散性合成并不局限于羟基异喹啉,常见的氮亲核试剂(如苯胺、N-甲基苯胺和N-烯丙基苯胺)也能实现这一转化,以中等至优良的e.e.值得到相应胺化产物的两种对映异构体。并且手性胺化产物很容易地进行一系列衍生化,具有潜在的合成潜力。
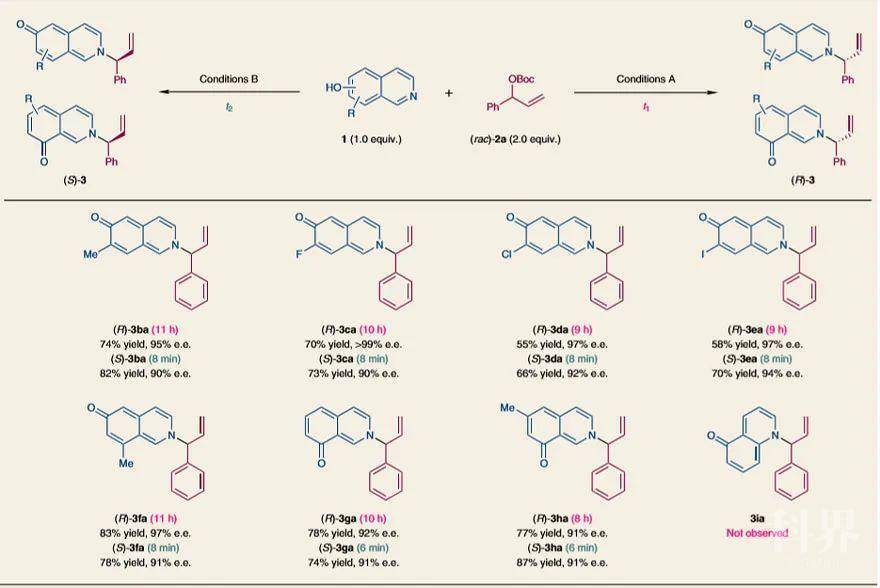
图5. 羟基异喹啉底物范围。图片来源:Nat. Chem.来源:X-molNews X一MOL资讯
原文链接:http://mp.weixin.qq.com/s?__biz=MzAwOTExNzg4Nw==&mid=2657646639&idx=1&sn=499c88194f565889707f7b4553bcf4fb&chksm=80f851ffb78fd8e994cdd4020261312e45b9a982c3dfad5f5d15956b2e8b5ff0550b0ece656b&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn






 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号