科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-09-27
近日,华西医院李为民、谢丹教授团队在cancer research上发表了基于非小细胞肺癌的多组学研究成果。Cancer Research(IF:8.378)是世界上创立最早、规模最大、享誉盛名的美国癌症研究协会会刊。该研究由四川大学华西医院独立完成,通讯作者为华西医院呼吸与危重医学科李为民教授和生物治疗国家重点实验室谢丹教授,本研究得到了国家自然科学基金等项目的支持。

图1、文章标题
肺癌是全世界肿瘤源性死亡的主要致死因素之一。由于其复杂的发病机理,目前为止没有有效的治愈方法。近年来,肺癌基因组测序研究已揭示部分基因突变与肺癌之间的关联,但仍有一部分肺癌患者没有明显的驱动基因突变,而是在表观遗传层面发生了改变。其中,染色质开放区域是一项重要的表观遗传学特征。开放染色质的存在往往意味着相应DNA调控元件的激活,从而调控下游基因表达。鉴于复杂的基因调控模式,单纯观测一个维度的数据改变已经无法很好地解析肿瘤的发生发展过程。因此,整合多组学数据(基因组,转库组,表观遗传组),从多个层面揭示肺癌复杂的基因调控网络具有重要意义。
该研究团队整合ATAC-seq技术、全基因组测序技术和转录组测序技术,研究肺癌基因调控关系。通过对50例非小细胞肺癌组织的染色质开放区域分析,研究者发现不同病理类型的肺癌具有各自的开放区域特征:如肺鳞癌特有的开放区域出现在上皮细胞角质化过程有关的基因周围;早期肺腺癌特有的开放区域出现在与预后有关的基因周围等。
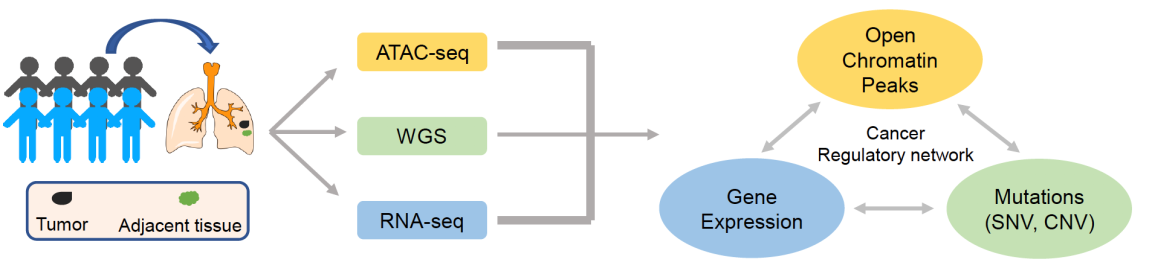
图2、研究设计
此外,研究者发现在肺癌样本中出现了一些较宽的开放区域分布(broad open chromatin peaks)。这些特殊的开放区域长度在1kb 到180kb之间,主要在基因的转录起始位点附近出现富集。它们不但表现出癌种特有的分布特征,还与基因表达的混乱程度相关。这些区域附近的肺癌驱动基因如EGFR、ERBB3等都在样本中表现出较大的表达量波动。该现象提示宽开放区域可能参与了肿瘤基因调控紊乱的过程。结合DNA测序中染色体结构变异数据,研究团队还发现宽开放区域在基因组拷贝数量增加的基因片段中出现富集。与此同时基因组拷贝数目变异(CNV)还与开放区域的信号及基因表达量有较强的关联性,即CNV会协同染色体开放区域影响基因表达调控。在这项研究中,研究团队观测到了大量受到这种表达调控模式影响的基因。
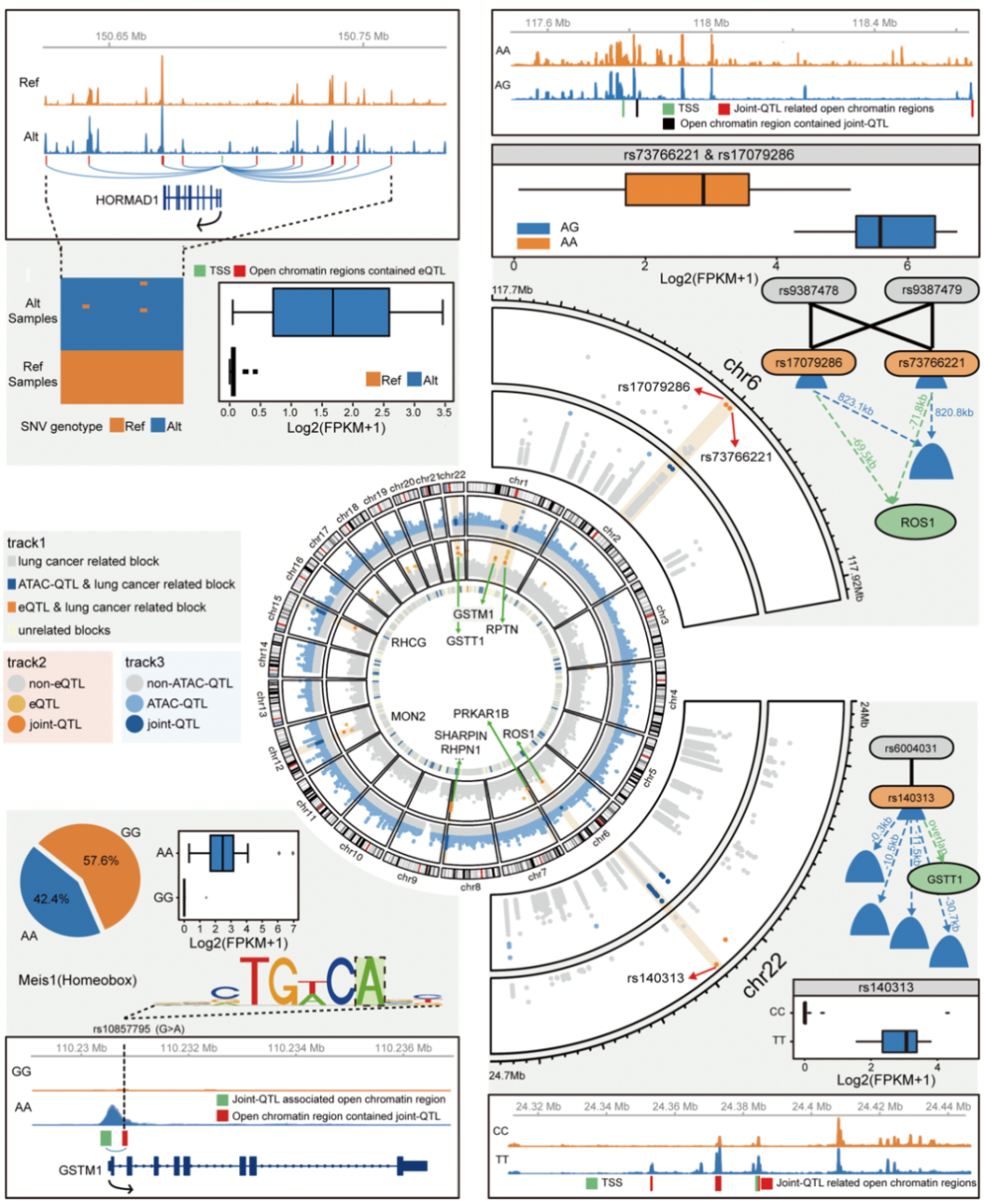
图3、多组学数据下的肺癌分子调控网络
多组学数据为解析肺癌基因调控模式提供了便利。在文章最后,研究团队通过分析整合不同组学信息之间的相关性,揭示了基因组序列变异、大片段DNA结构改变、开放染色体区域与基因表达改变之间复杂的分子调控网络。这种调控模式不但验证了部分先前肺癌的全基因组关联分析研究(GWAS)的结果,包括与ROS、GSTT1基因表达调控具有相关性的一些SNP位点。研究团队还从中发现了大量位于非编码区域的基因组区段在调控网络中发挥着重要功能,结合生物信息学手段预测了这些原先被认为是“暗物质”的DNA片段对基因进行调控的模式以及相关转录调控因子,并通过肺癌细胞系实验针对一部分位点的功能进行了验证。
本研究整合多组学数据,揭示了非小细胞肺癌基因组调控网络,并且可利用前沿的基因组学方法来鉴别不同亚型的肺癌,发现了21个与基因组开放区域及转录本表达调控有关的数量性状座位(joint-QTLs),并且进一步证实了GWAS研究中所提出的与肺癌风险相关的位点参与基因组调控网络的模式。这些位于非编码区域的调控位点,可以作为肺癌诊断以及药物设计的靶标,指导肺癌的精准化治疗。
原文链接:
https://cancerres.aacrjournals.org/content/early/2019/08/08/0008-5472.CAN-18-3663.full-text.pdf
环境友好的农林害虫生态调控与生物防治技术——2017高级研修班总结

合肥研究院发现环形RNA调控小细胞肺癌的关键信号通路

肺癌大咖齐聚线上 共话肺癌精准诊疗|肺癌精准诊疗学术论坛精彩回顾

【学术前沿】JMCB | 惠静毅团队揭示RNA结合蛋白QKI调控肺癌关键可变剪接事件
昆明动物所等发现Gemin6在非小细胞肺癌中的调控机理

JMCB:惠静毅团队揭示RNA结合蛋白QKI调控肺癌关键可变剪接事件
国际肺癌日 | 如何有效预防肺癌?这些知识你该知道
【人文】肺癌术后和肺癌患者哪些可以接种新冠疫苗
Molecular Plant 出版“植物激素”专辑

Nat Comm:钟波/褚倩研究组发现非小细胞肺癌免疫治疗调控的新机制


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号