科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-10-18
来源:Deep Omics
文章题目:Rare variant phasing using paired tumor:normal sequence data
关键词:肿瘤基因组学、VAF、生殖系SNV、单倍体分型
本文提出了基于肿瘤和正常的配对测序数据进行单碱基变异(SNV)的单倍体分型方法。该方法利用在肿瘤细胞中由拷贝数变化(copy number alteration, CNA)导致的SNV等位基因频率(variant allele frequency,VAF)改变来解析生殖系(germline)SNV单倍体。该方法使用在TCGA数据库的6180个样本上,得到了与其他常用单倍体分型方法高度一致的结果。而且,该方法比其他基于测序片段连通性的方法可以多分型33%以上的位点。

VAF单体型分型方法介绍
VAF单倍体分型方法基于一个基础并直观的事实。图1A和图1B分别是涉及到的两种不同的情况。图1A中,若正常组织中两个生殖系SNV位点(突变A和G)分别处于同源染色体的两条不同染色体上(即两个单倍体),则当其中某一个片段(或整条染色体)在肿瘤样本中发生拷贝数增加的时候,处于拷贝的片段(或染色体)上的SNV的VAF会随之发生增加,如图1A的突变A。同时,因处于不同的染色体上,另一个SNV位点的突变G的VAF会降低。同理,若两个SNV位点处于同一单倍体上,则两个突变的VAF改变是同步的,即同时增加或减少(图1B)。因此,在肿瘤样本中若发现同一染色体的邻近的不同SNV位点VAF变化是相反的,则表明这两个SNV是不同单倍体,称之为反式分型;若VAF变化是相同的,则表明这两个SNV处于同一个单倍体上,称之为顺式分型。
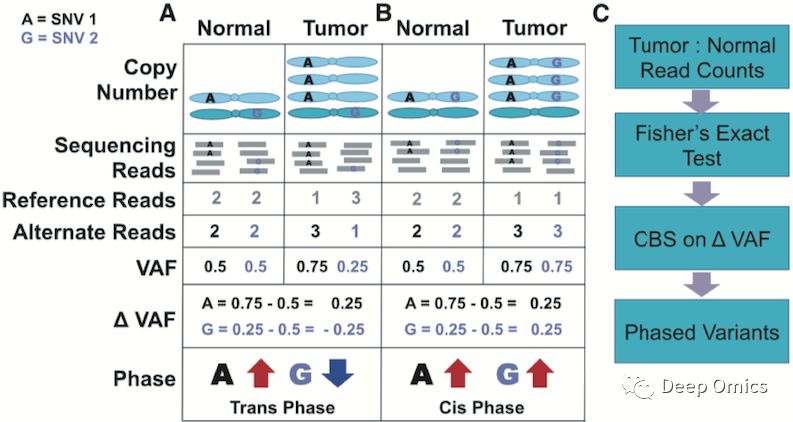
图1 VAF单倍体分型方法示例及主要步骤。
基于以上现象,可以通过肿瘤细胞中等位基因频率相较于正常细胞的改变,对生殖系SNV进行单倍体分型。但在做分型前,必须保证以下两点:
分型到同一个单体型的SNV位点需要处于同一个发生拷贝数增加或减少的DNA片段上,即同一个SCNA(Somatic Copy Number Alteration)片段上。
每个参与分型的SNV位点的VAF在正常和肿瘤细胞之间存在显著变化。
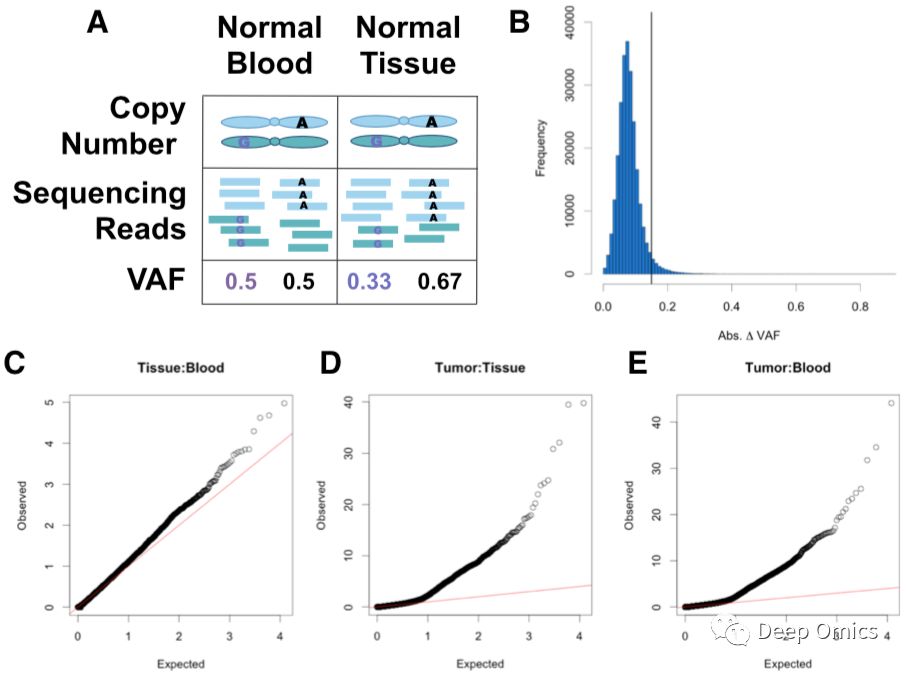
图2 采用血液和正常组织均有测序的样本来鉴别SCNA及确定阈值。
对于第一点,研究人员认为,在同一个SCNA上,各SNV位点的VAF变化绝对值应该是连续且相近的。基于此,研究人员对VAF变化绝对值使用循环二分法(circular binary segmentation,CBS)进行SCNA片段的确定。因为VAF变化绝对值有一定的误差,并且此误差来自于测序序列采样,这种情况下VAF变化并不是由SCNA导致的(图2A)。考虑到这种误差,研究人员使用TCGA中同时有血液和正常组织外显子测序的416例样本做了CBS分段并对分出的249,471个片段的VAF变化绝对值的平均值做了统计(图2B)。结果显示,95%的片段的VAF变化绝对值小于0.14。因为血液和正常组织中理论上是没有SCNA出现的,研究人员使用0.14为判断SCNA的临界值,即在5%的错误率下,片段中SNV位点的VAF变化绝对值的平均值大于或等于0.14时,该片段被判断为SCNA。
对于第二点,研究人员利用SNV位点的等位基因测序深度做了假设验证(Fisher’s exact test),以判断每个SNV位点是否在统计学上具有显著的VAF变化。只有拥有显著p值的位点才会被考虑进行单体型分型。再次使用416例血液和正常组织的外显子测序数据,研究人员发现对生殖系SNV位点做假设验证的p值遵循了他们期望的分布,p值小于0.05的杂合SNV位点所占比例的中位数为6%(图2C)。然而在肿瘤与对照组织(血液或正常组织)比较中,中位数则为17%(图2D和2E)。
结合使用 ①VAF变化绝对值的临界值来判断SCNA片段 和 ②对位点做假设验证判断VAF是否有显著变化,研究人员根据VAF的增加和减少对生殖系SNV进行单倍体分型(图1C)。假阳性方面,该方法在416例血液和正常组织的外显子测序数据上,错误地纳入单体型分型的生殖系SNV位点仅占0.3%。
VAF单体型分型方法与常用分型方法的比较(TCGA数据及10x测序)
研究人员将VAF分型方法在6180个TCGA样本中进行了测试,并将分型结果与HapCUT2、phASER和SHAPEIT的结果进行比较(图3)。结果表明,在HapCUT2和phASER结果相同的位点中,VAF分型方法达到了很高一致性(99%);而在HapCUT2或phASER独有的位点中,平均有9%的被VAF分型方法成功解析。研究人员发现,将本方法与HapCUT2和phASER结合使用进行单倍体分型时,能很大程度上增加分型位点(平均33%,图3A),并且对HapCUT2和phASER不能作出分型的位点,VAF分型方法平均成功解析了942个(图3B)。
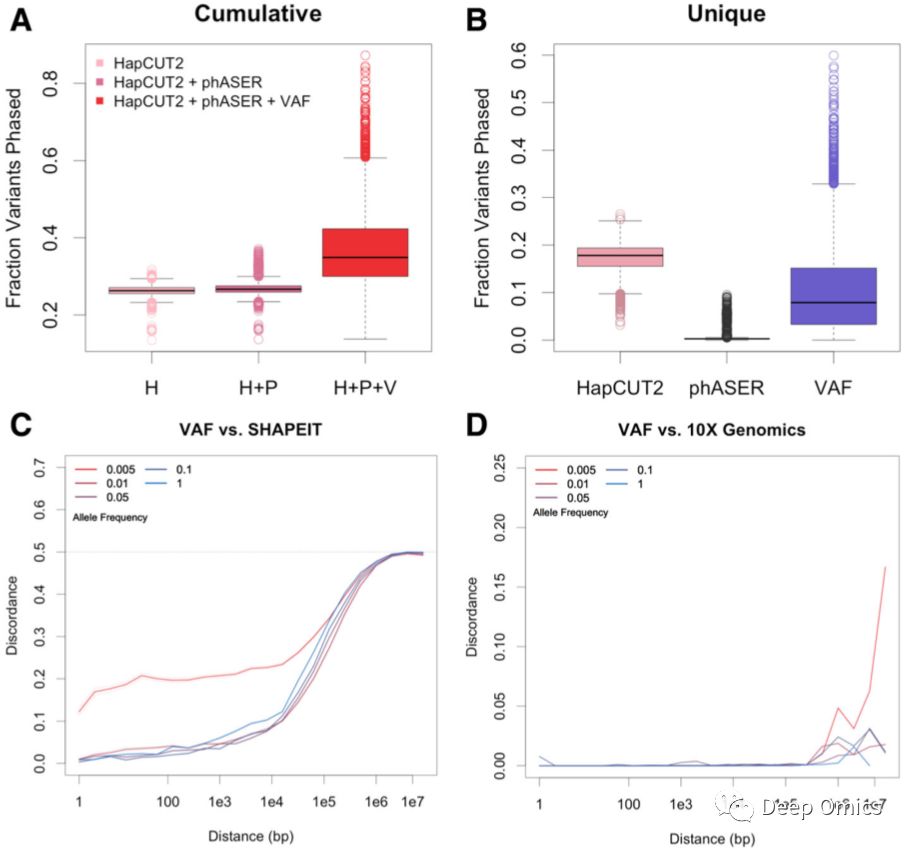
图3 多种分型方法的结果比较。
研究人员在长距离分型上将VAF方法和SHAPEIT做了比较,并统计了不同长度下两种方法分型结果的一致性(图3C)。在10kb的长度内,VAF方法和SHAPEIT有很高的一致性(>90%)。大于10kb的长度时,两者结果一致性随长度增加而降低。同时,对不常见的SNV变异位点(人群等位基因频率小于0.005),VAF和SHAPEIT的不一致性较高。这是因为SHAPEIT使用人群数据库对个体进行分型时,数据库信息权重会远远超过个体信息。在低人群等位基因频率的情况下,导致结果在个体水平的不准确。同样地,研究人员用COLO829细胞系比较了VAF与10x barcode测序的分型结果,整体一致性达到了99.23%,并且区域长度(<1Mb)和人群等位基因频率对结果几乎没有影响(图3D)。
我们的思考
本文提出了以肿瘤和正常配对样本为基础,利用由SCNA导致的等位基因频率的变化来进行生殖系SNV位点的单倍体分型。与常用的单倍体分型方法(如SHAPEIT,phASER,LongRanger)相比,这种方法不直接借助于测序序列的连通性,也不采用人群单倍体数据库作为参考,消除了来自人群的潜在噪音,并提高了个体单倍体分型的位点数目和准确度。
使用正常配对的血液和组织样本,研究人员建立了一个等位基因频率变化的背景分布,以判断SNV的VAF变化的程度,从而确定SCNA片段,确保分型的准确率。这种通过实际采样进行背景评估的思路值得借鉴。
由于VAF单倍体分型方法依赖于SCNA的存在,并且必须使用肿瘤样本和配对的正常样本,这是其在应用上的一个限制。这说明了,该方法是常用单倍体分型方法在分析肿瘤数据时的一个重要补充。
来源:gh_cc101106ed81 Deep Omics
原文链接:http://mp.weixin.qq.com/s?__biz=MzI2NDg2MjgyOA==&mid=2247483671&idx=1&sn=a01055baecbb0fa8ac70b2565d4a8343&chksm=eaa76c14ddd0e502859c350e1a85413f46e41cce4bec47944c3c351f2befa8d1ce33add18963&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

背靠背!华中农大/扬州大学分别揭示了稻米好看又好吃的等位基因

JCI:等位基因特异性RNA干扰有望治疗腓骨肌萎缩症2D型
先祖性墨西哥类蜀黍等位基因可导致更高产的密集种植玉米

扬州大学刘巧泉课题组克隆了协调稻米食味和外观品质的稀有Wx等位基因

基因编辑婴儿:已经出了什么问题?还可能会出什么问题?
昆明动物所等在等位基因特异的RNA编辑研究中取得进展

杂合的等位基因,诱导小麦单倍体效率更高

新的等位基因让水稻更好吃

Nuc Acid Res:单细胞水平揭示等位基因对雌激素的反应性

Nature发表基于选择性非同源末端连接修复的基因治疗方法


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号