科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-10-19
来源:药学进展
[摘要] 别构药物具有高选择性和抗耐药性的优势。相对于正构药物较为成熟的研究策略,目前别构调控分子的发现存在很大挑战。总结笔者实验室在别构调控研究领域取得的进展,包括别构位点的理论计算预测方法、别构调控分子理性设计的成功示例,并讨论了别构药物研发领域面临的挑战以及新的发展动向。
别构(又称“变构”)调控(allosteric regulation)是指效应分子结合在蛋白质三维结构上不同于底物位点的别构位点(allosteric site),通过多种信号传导途径而导致蛋白质活性改变的过程。作为一种细胞内有效的调控蛋白质功能的方式,别构调控在很多的生物过程中发挥着重要作用,包括酶催化、信号传导、转录调控 、代谢调控、协同进化等。自 50 多年前提出别构效应以来,除了别构调控的详细机制尚未研究清楚,别构概念、实验和理论研究都有很大进展。别构调控在蛋白质中广泛存在,因此针对别构位点进行药物设计为控制蛋白质活性提供了另外一条途径。药物根据其作用机制可以分为正构药物(orthosteric drug)和别构药物(allosteric drug)。
正构药物结合在蛋白质的活性位点,占据底物或辅基的结合位点,通过阻碍底物或者辅基与蛋白质的结合而抑制蛋白质的活性;别构药物则通过结合在非活性位点来改变或者阻断催化或者底物的结合来发挥作用。目前除了在美国上市的18种别构调控剂,大部分已上市药物为正构药物。相对于正构药物,别构药物有着如下优势:1)选择性高。正构位点药物,由于其同家族蛋白活性位点的保守性,存在选择性的问题;别构药物则由于结合位点的结构保守性低,因而副作用更小。2)别构药物和底物可以同时发挥作用,其调控功能具有上限性,毒性小。3)别构位点提供了新的结合位点。从功能上讲,别构药物既可以下调蛋白的活性成为抑制剂,也可以上调蛋白活性成为激活剂。目前已有多种针对不同靶标的别构调控分子被发现,包括 G 蛋白偶联受体、离子通道、酶以及其他的靶标,然而大部分别构调控分子还是通过高通量实验筛选获得 ,别构调控分子的理性设计和发现仍颇具挑战性。
近年来有关别构调控分子的理性设计和发现取得了令人鼓舞的成果,详见近期综述。笔者实验室对于别构调控的研究源于对于人类花生四烯酸(AA)代谢网络调控的研究,为了有效控制炎性分子的产生,需要上调网络中的一些靶标,而酶的激活只能通过别构调控来实现 ,笔者实验室发展了别构位点预测方法和别构调控分子的理性设计方法,并在一些重要靶标上获得了上调的别构分子。本文将主要介绍别构位点的预测与确认,以及别构调控分子的发现实例。
1 基于理论计算的别构位点发现
理性设计别构调控分子,首先需要寻找靶标的别构位点(见图 1)。虽然原则上所有的蛋白质都是别构的,但是被实验确认的别构位点数量有限,目前最大的别构蛋白数据库(allosteric database,ASD)也只收录了 1 949 个蛋白质。近十几年来,很多研究者也一直在关注如何发现潜在的别构口袋。发现别构口袋的方法可以分为实验(包括高通量筛选、点突变、核磁驰豫扩散实验和氘氢交换质谱等)和理论计算两大类。单纯开展实验工作的研究,一方面工作量很大,另一方面实验发现具有偶然性;而理论计算方法在降低成本的同时可以大幅提高别构位点发现的成功率,有利于别构抑制剂的机制的探究,因此,需要发展基于理论计算的别构位点预测方法。
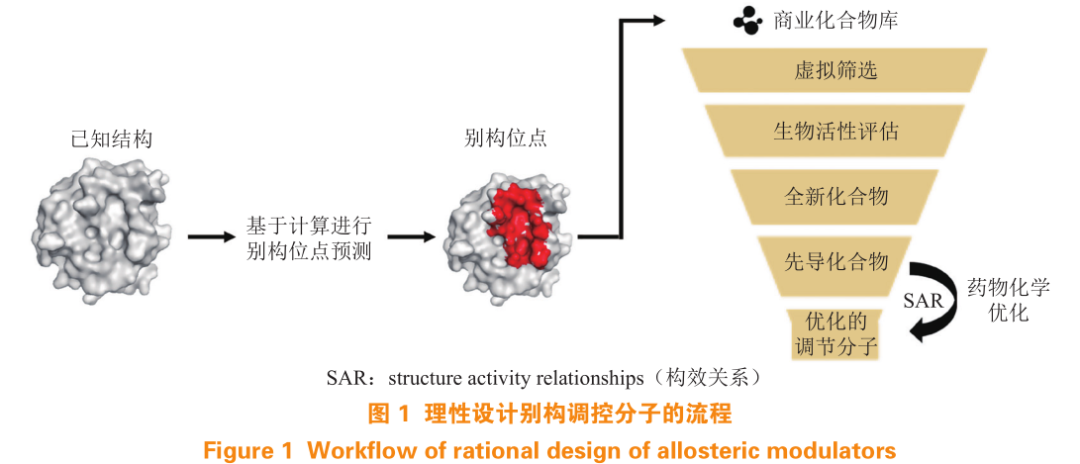
当已知蛋白质活性和非活性 2 种状态的结构时,可以根据这 2 种结构的转变情况进行别构位点预测。Qi 等和 Ma 等发展了利用粗粒化二态Gō 模型预测蛋白质别构口袋的方法。首先用二态Gō 产生蛋白质活性构象和非活性构象的平衡系综,然后在探测到的蛋白质表面可成药结合位点(非活性位点)内,用微扰的方法改变残基之间的相互作用模拟小分子的结合过程。若微扰的施加导致蛋白质构象系综发生变化,那么此微扰施加的位点便可能是一个潜在的别构位点。利用这种方法,结合笔者实验室发展的口袋探测程序 CAVITY ,成功发现了大肠埃希菌中 D-3- 磷酸甘油酸脱氢酶(E.coli D-3-phosphoglycerate dehydrogenase,PGDH) 的 新的别构位点,并在此基础上经计算虚拟筛选和实验获得了别构抑制剂。这种预测蛋白质别构口袋的方法需要已知所研究蛋白质的 2 种构象及活性有差别的状态,故其应用范围有局限性。
大多数情况下并不知道所研究的蛋白质是否会被别构调控,更没有调控后的结构数据可用。针对这种情况,Ma 等假设蛋白质正构位点和别构位点运动上的强相关性是一种普遍存在的现象,可用于别构位点预测,并采用粗粒化的高斯网络模型对这种相关性进行了研究。他们在测试 23 个聚集状态为单体的别构蛋白质的 24 个已知别构口袋后发现,其中 23 个已知别构口袋与其对应的正构口袋在运动上显示了强相关性。随后他们又分析了几种多聚体别构蛋白质,发现其正构口袋与别构口袋也有很强的相关性。并且这种结果不依赖于蛋白质的初始结构,具有鲁棒性。基于这些结果开发了一种在已知一种蛋白质构象的情况下,可以快速预测蛋白质别构口袋的理论计算方法和程序 CorrSite。
为 方 便 用 户 使 用, 以 上 的 程 序 CAVITY 和CorrSite 已集成在 CavityPlus 上。CavityPlus 是一个 Web 在线计算平台,使用蛋白质三维结构信息作为输入,提供蛋白质口袋检测和各种功能分析。该平台应用 CAVITY 来检测给定蛋白质结构表面上的潜在结合位点,并根据配体性和可成药性评分对其进行排名。可以使用 3 个子模块 CavPharmer 、CorrSite 和 CovCys 进一步分析这些潜在的结合位点:CavPharmer 使用基于受体的药效团建模程序 Pocket 以自动提取口袋内的药效团特征;CorrSite可以识别潜在的别构配体结合位点;CovCys 则能自动检测可药用的半胱氨酸残基,可在设计共价别构分子时用于寻找合适的共价结合位点。
其他研究团队也通过不同计算策略开发出多种类型的别构位点预测方法。在这些预测方法中,笔者实验室开发的 CorrSite 表现出其优越性:与基于二态 Gō 模型预测别构口袋的方法相比,CorrSite 只需蛋白质任意一种活性状态的结构即可,使用范围更广;与利用分子动力学(moleculardynamics,MD) 模拟来预测别构口袋的方法相比,CorrSite 耗时很短,使用方便;与仅基于结构的方法相比,CorrSite 考虑到蛋白质结构中各个部分的相互作用;与同样使用正则模式分析(normal mode analysis,NMA)来鉴定蛋白质别构位点的 PARS(Protein Allosteric and Regulatory Sites)方法相比,CorrSite 的算法更为简单,正确率也更高。不过,受限于当时可用的蛋白数量以及计算量,CorrSite存在一定的局限性,这也是进行别构位点预测研究遇到的共性的问题。随着已知别构蛋白质复合物结构数量的增加,CorrSite 可以在更大的数据集上进行验证和优化。
目前,基于蛋白质动力学的别构位点研究受到很多关注,例如结合口袋特征及 NMA 分析进行蛋白质别构位点预测的方法 AllositePro 表现出较好的预测效果;Yu 等研究了计算原子运动相关性时的角度问题;Zhang 等在 2019 年发表的一篇综述中提出蛋白质在进化过程中利用蛋白质内在动力学来调控别构与正构功能。基于蛋白质动力学相关性的别构位点预测方法,在多个体系的别构调控分子设计中得到了成功应用。
2 别构调控分子的理性设计实例
在找到靶标的别构位点之后,通过计算与实验验证相结合的方法有望发现别构调控分子。下面介绍笔者实验室近年的研究进展。
2.1 别构激活剂的发现
与抑制剂的发现相比,蛋白质激活剂的发现更加困难,难点一在于很难通过计算预测别构分子与靶蛋白结合后起上调或下调的作用;难点二在于激活剂往往使得蛋白质处于能量较高、更加动态的活性构象,因此不能稳定在单一构象的状态,这使得激活剂与靶蛋白的复合物晶体结构解析更为困难。目前,发现激活剂的主要途径依旧是高通量筛选,也有一些理性设计激活剂的例子被报道,如 SIRT6激活剂的发现等,尚无普遍适用的开发激活剂的方法。
2.1.1 磷脂氢谷胱甘肽过氧化物酶激活剂 磷脂氢谷胱甘 肽 过 氧 化 物 酶(glutathione peroxidase 4,GPx4),是一个含有硒的谷胱甘肽过氧化物酶。GPx4 与细胞膜修复、炎症及铁死亡抑制等诸多生物调控过程相关 。上调 GPx4 的活性对于炎症以及铁死亡相关的疾病治疗有益。因此,开发GPx4 激活剂对于相关的疾病治疗具有重要意义。
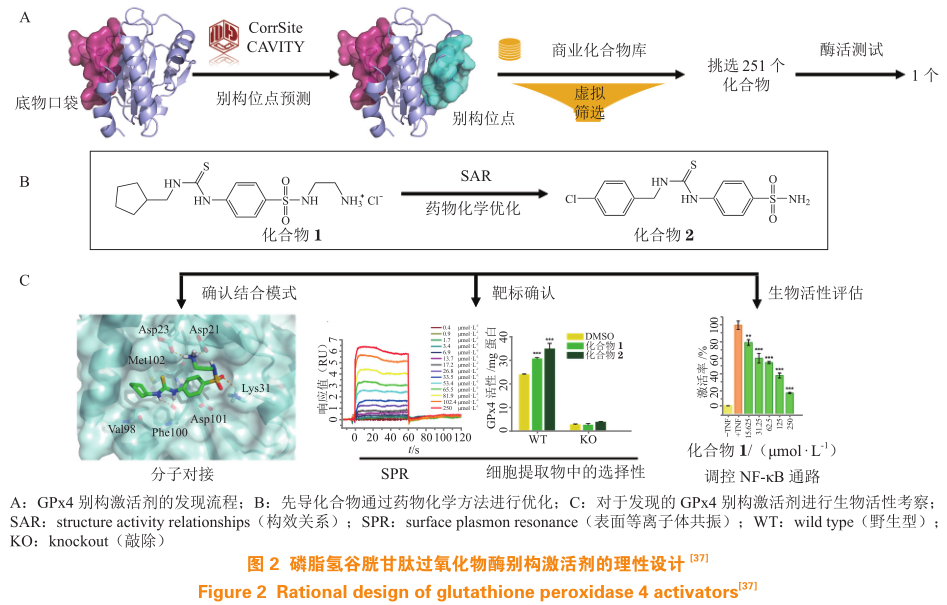
Li 等使用 CorrSite 和 CAVITY 在 GPx4的三维结构上成功发现了一个新的别构位点(见图2A)。预测的别构位点 Cavity 打分最高且 CorrSite得分为 1.5(CorrSite 方法中通常将 CorrSite 得分大于 0.5 的口袋预测为潜在的别构位点)。该别构位点处于底物位点的背面,由 3 个酸性残基(Asp21、Asp23 和 Asp101)、2 个 碱 性 残 基(Lys31 和Lys90)和 7 个非极性残基(Ile22、Ala93、Ala94、Val98、Phe100、Met102 和 Phe103) 包 围。通 过对 SPECS 化合物库进行计算虚拟筛选挑选后购买的 251 个化合物进行酶活测试,最终得到激活剂分子 PKUMDL-LC-001(1)。化合物 1 以剂量依赖性方式将人源 GPx4 的 U46C 突变体(human GPx4 U46C mutant,hGPx4-C)的酶活性提高到原始酶活性的 263%,其与 hGPx4-C 的平衡解离常数(K D )为(63±5)mol · L -1 。对该化合物进行结构修饰后,得到活性提高的化合物 1d4(2,见图 2B),其达到 150% 激活所需的浓度为 20 mol · L -1 。突变实验中,将别构口袋中的关键氨基酸 Asp21 和 Asp23 突变后测试发现,化合物 1 和 2 对 2 个单突变(D21A和 D23A)和 1 个双突变(D21A/D23A)激活活性降低,证实化合物 1 和 2 结合在所预测的别构位点上(见图 2C);敲除 GPx4 后,化合物 1 和 2 对于细胞提取物的激活效果消失,证明其在细胞中的特异性。化合物 1 对于铁死亡的抑制、对于 AA 代谢网络中炎症前脂质中间体的抑制以及对于核因子 κB(nuclear factor kappa B,NF-κB)通路的抑制均进一步证实其作用于 GPx4。
2.1.2 15-脂氧合酶激活剂 人网状细胞 15-脂氧合酶(15-lipoxygenase,15-LOX)是相对分子质量为75 000 的脂肪酸双加氧酶,其产物 15-羟基二十碳四烯酸(15-HETE)可以进一步转换为具有抗炎活性和消炎能力的脂蛋白(lipoxins,LXs) 。上调15-LOX 可以促进抗炎因子生成且同时减少促炎因子 ,是潜在的治疗炎症相关疾病的靶点。另外,15-LOX 与癌症和铁死亡密切相关。因此,针对 15-LOX 设计的激活剂,既能够作为分子探针工具研究其功能也能够开发为治疗相关疾病的药物。
Meng 等应用 MutInf 方法寻找与 15-LOX 底物结合位点二面角运动相关性高的残基,并结合CAVITY 判断这些残基是否适于形成可成药口袋,成功地在 15-LOX 底物口袋附近找到一个新的别构位点 cavity 1。随后,笔者实验室使用 CorrSite 对15-LOX 所有的潜在别构口袋进行了预测,cavity 1的 CorrSite 得分为 1.1,排名第三。与 MutInf 方法相比,CorrSite 方法耗时少且使用方便。
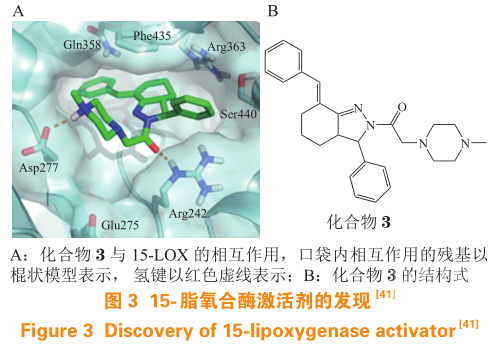
笔者实验室通过对 cavity 1 使用 SPECS 化合物库进行计算虚拟筛选和实验研究,得到 1 个激活剂和 11个抑制剂分子,其中激活剂分子 PKUMDL_MH_1001(3)的半数激活常数(AC 50 )为(6.8± 0.4) mol · L -1 ,K D 为(3.9±0.1)mol · L -1 。虚拟筛选后对化合物进行活性测试,获得了另外 2 个激活剂分子。将激活剂与抑制剂化合物的结构进行比对后发现,激活剂分子中均包含可以与带负电残基形成强氢键的正电性基团。随后,笔者实验室对 15-LOX 的别构激活及抑制机制进行了研究,发现激活剂与 Asp277 形成氢键后可以稳定 15-LOX 活性构象,减少底物自抑制的作用;而抑制剂倾向于使活性位点口袋的“开合运动”受到阻碍。突变实验表明化合物 3 对于15-LOX 的突变体 R242A 和 D277A 的激活活性大幅下降,验证了化合物3结合在所预测的别构位点(见图 3)。此外,在人的中性粒细胞、人全血和小鼠腹膜炎模型中,化合物 3 不仅增加了 15-LOX 的产物 15-HETE 水平,还减少了促炎性介质的产生。更有意义的是,化合物 3 与 5-LOX 或 COX 抑制剂的联合使用可平衡 AA 代谢网络中的不同途径,这为阻断炎症的新策略提供了依据。
2.2 别构抑制剂的发现
2.2.1 D-3-磷酸甘油酸脱氢酶别构抑制剂 D-3-磷酸甘油酸脱氢酶(D-3-phosphoglycerate dehydrogenase,PHGDH)催化生物体内合成丝氨酸的第一步反应,是丝氨酸合成的决速步骤。PHGDH 在癌症的发展中起着重要作用;PHGDH 的基因敲除或使用PHGDH 的小分子抑制剂都会大幅抑制对 PHGDH敏感的细胞在体内或体外的生长,对 PHGDH 不敏感的细胞系受到的影响则较小。靶向 PHGDH 的抑制剂有着广泛的应用前景。由于 PHGDH 的底物口袋小,辅基NAD + 在细胞中的浓度达到毫摩尔级,正构位点的抑制剂面临选择性差而无体内活性的问题。因此,针对 PHGDH 设计别构抑制剂是一种有效的策略。
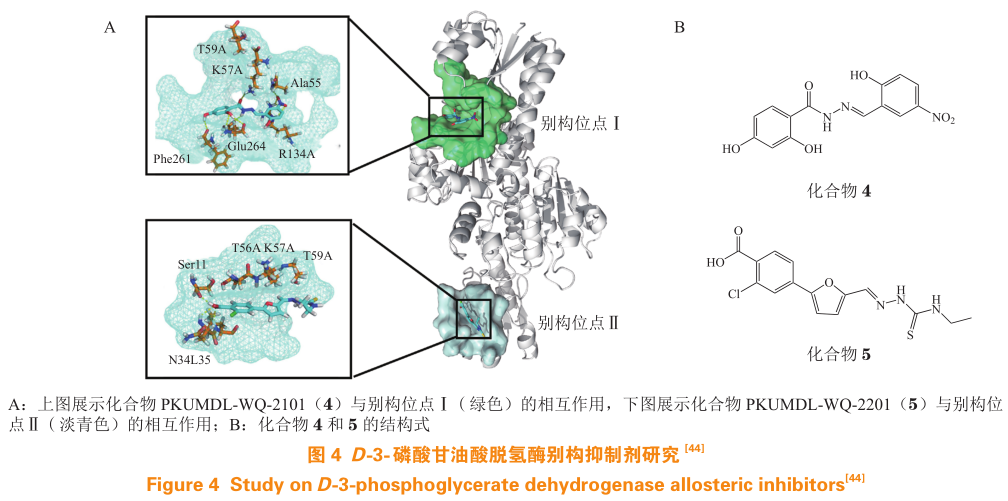
考虑到 PHGDH 的全长结构尚未解出,Wang等利用现有的仅包含 2 个结构域的晶体结构(Pdb code:2g76)使用 CAVITY 程序进行了别构口袋的预测,并获得了 2 个别构口袋(位点Ⅰ和位点Ⅱ,见图 4)。位点Ⅰ靠近活性位点和 NAD + /NADH 辅因子结合位点,位点Ⅱ靠近底物结合域。随后,笔者实验室对 SPECS 化合物库进行了计算虚拟筛选,得到 1 个结合在位点Ⅰ的化合物 PKUMDL-WQ-2101(4)和 3 个结合在位点Ⅱ的化合物:PKUMDL-WQ-2201(5)、PKUMDL-WQ-2202、PKUMDL-WQ-2203。其中活性最好的化合物 4 和 5对 PHGDH 的 IC 50 为(34.8±3.6) 和(35.7±8.6 )mol · L-1 ,二者与 PHGDH 的 K D 分别为(0.56±0.10)和(66.9±1.9) mol · L -1。突变实验中,化合物 4 对于突变位置在别构位点Ⅰ的 PHGDH 突变体 R134A和 K57AT59A,化合物 5 对于对于突变位置在别构位点Ⅱ的 PHGDH 突变体 T59A 和 T56AK57A 的抑制活性均大幅降低,验证了化合物 4 和 5 分别结合在别构位点Ⅰ和位点Ⅱ。在细胞实验中,化合物4 和 5 对 PHGDH 敏感性细胞系 MDA-MB-468 的EC 50 分别为 7.7 和 6.9 mol · L -1 ,同时对 PHGDH 不敏感性细胞系呈现出很好的选择性,表明在细胞中化合物 4 和 5 靶向作用于 PHGDH。研究发现,这2 个化合物在 PHGDH 敲除后的细胞中均无特异性活性;二者还减少了丝氨酸合成通路及其下游通路的代谢产物的量,与PHGDH基因敲除的影响类似。此外,二者在动物实验中也展现出了对于肿瘤组织的抑制效果。
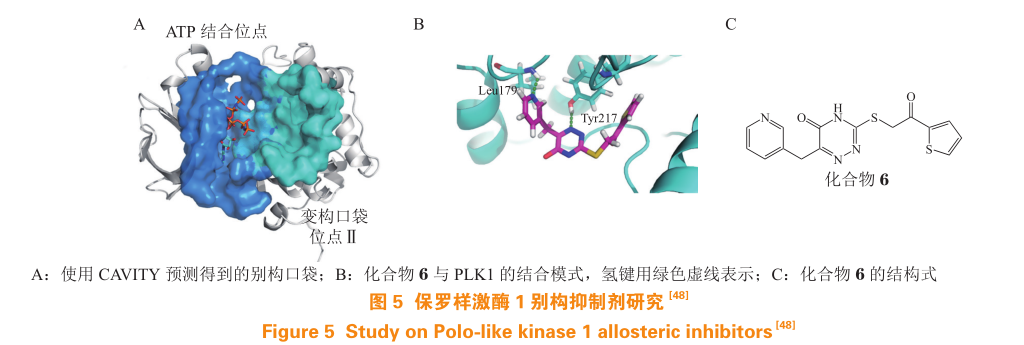
2.2.2 保罗样激酶 1 的非 ATP 竞争型抑制剂 保罗样激酶 1(Polo-like kinase 1,PLK1)是哺乳动物细胞中参与多步有丝分裂的赖氨酸/苏氨酸激酶,其结构包括识别底物的调控结构域(Polo-box domain,PBD)和催化底物的激酶结构域(kinase domain,KD)。研究表明人源 PLK1 在多种恶性肿瘤中过表达 ,抑制 PLK1 可以抑制癌细胞的生长,因此,PLK1 被认为是一个潜在的抗肿瘤靶标。已有的 PLK1 抑制剂主要作用于 KD 的 ATP 结合口袋,是 ATP 竞争型抑制剂。该类抑制剂活性高但选择性低,作为药物使用产生的不良反应较多,而非 ATP竞争型抑制剂能够提高选择性以及延缓突变。Yun 等使用 CAVITY 对 PLK1 晶体结构进行了可成药性口袋分析计算,在 ATP 结合位点的附近发现了全新的别构口袋(见图 5A)。通过对别构口袋中的位点Ⅱ进行虚拟筛选并进行 PLK1 体外酶活性测试,发现了多个活性化合物。其中活性最好的化合物 6 对全长 PLK1 酶活的 IC 50 为(13.1± 1.7)mol · L -1,与 ATP 没有竞争性结合(见图 5B)。
3 结论和展望
笔者实验室开发了预测别构位点的方法,预测了 GPx4、15-LOX、PHGDH 和 PLK1 中的别构位点,并分别针对这些位点成功发现了别构调控分子。
在过去的几十年里,基于结构的药物设计大大加速了药物发现的进程。别构药物相对于正构药物所具有的高选择性和抗耐药性受到很多关注。越来越多的别构药物正被发现,别构药物涉及的靶标也从最初的 G 蛋白偶联受体和激酶扩展到现在的 14种蛋白类别,也有不少别构调控分子被发现。尽管有这些成功的例子,但是基于结构的别构药物设计依然具有很大的挑战性。主要在于以下 3 个方面:
1)别构位点的预测。其一,目前有很多方法和软件用于别构位点预测,但因已知的适合用来训练和测试的别构蛋白的样本数量及计算量有限,这些预测方法本身的精度及适用性存在差异,还需要其他更多的研究来证明其有效性。其二,选择什么样的构象开始进行预测是研究人员在进行别构药物设计时遇到的首要问题。最近,张健研究组发现,当蛋白质中正构和变构位点之间的距离小于 5 Å 时,选择结合底物的蛋白构象作为基于结构的变构药物筛选的初始输入,有助于发现变构药物。本文介绍的例子中,2 个例子(PHGDH 和 PLK1)使用结合底物的蛋白构象作为初始输入,而预测得到的别构位点都在活性位点附近,支持了此结论。其三,在别构药物设计中,有些别构口袋在 apo(不结合别构药物的构象)状态时不存在(hidden allosteric site),这种情况下可能需要进行大规模分子动力学模拟,结合马尔科夫状态模型(Markov state model)以获得潜在的别构位点。
2)别构药物的优化(hit-to-lead optimization)。首先,别构药物的活性往往低于正构药物。由于别构药物的活性与其亲和力并无直接关系,用传统的方法对别构药物优化时,虽然可以提升药物分子的亲和力,但并不一定提升活性,因此产生“flat SAR”或者“limited SAR”的现象。其次,别构调控分子细微的差别即可能导致发挥的功能不同,理解别构分子的机制对于别构药物优化非常重要。例如,笔者实验室的 Bi 等在大肠埃希菌趋化研究中发现的化学效应分子(chemo effectors)类似物CHDCA 和 cis-PDA,二者分别是大肠埃希菌 Tar 受体的拮抗剂(antagonist)和引诱剂(attractant),其结构差别只是后者多了一个 N - H 基团,通过与Tar 受体的相互作用比较发现,引诱剂都是在合适的位置(靠近 Tyr149 和 Gln152)有 1 个氢键供体基团作为信号的触发基团。因此,别构调控分子发挥的作用可以分为两部分:一部分用于与受体结合;另一部分起到信号触发(trigger)的作用。类似的情况也在 15-LOX 激活剂的介绍部分提到过,即当结合在别构位点的分子没有带正电时成为抑制剂,而当带正电时就成为激活剂。综上,别构分子的优化复杂且富有挑战性。
3)相对于正构位点,别构位点的成药性往往更差,别构位点计算虚拟筛选的成功率也远低于正构位点,从而限制了别构药物的多样性。
别构药物的发展面临着种种挑战,但是其在一些新的方向越来越受到关注。比如,开发别构共价药物,可以保留别构药物的优势,同时也可以增加药物-靶标配体的半衰期从而降低毒性。笔者实验室所开发的 CovCys 程序可以探测蛋白质中能够形成共价的半胱氨酸,该程序也已集成在CavityPlus 中供直接使用。另外,在开发别构药物对于蛋白-蛋白相互作用界面的调控,不可成药靶标的调控,通过别构研究老药新用以及理解中草药在治疗复杂疾病时所发挥的作用等方面也有进展。从系统生物学及别构网络(allo-network)的角度进行别构药物的设计将会影响未来的药物发现和发展。
来源:ppsyxjz 药学进展
原文链接:http://mp.weixin.qq.com/s?__biz=MzA5MDY3ODExNQ==&mid=2651314866&idx=1&sn=7798ac1052492986b90b21f814b4059a&chksm=8bf48cbebc8305a87f6eb0aac442ef8476c32b03b6a8755085767af82b5e90044e0e4d282da4&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

山东大学药学院发现新一代高效抗耐药性HIV-1抑制剂

抗疟疾神药青蒿素的耐药机制终于被发现,为解决耐药性指明方向
山东大学刘新泳教授团队在抗耐药性抗艾滋病药物领域取得新进展
干预DNA损伤修复机制的抗耐药性四价铂抗癌前药
Science重磅:抗疟疾神药青蒿素的耐药机制终于被发现,为解决耐药性指明方向

2019冠状病毒病大流行增加了抗微生物药物耐药性的威胁

Science重磅:抗疟疾神药青蒿素的耐药机制终于被发现,为解决耐药性指明方向


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号