科技工作者之家
科界APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2019-11-15
来源:药学进展
中国药科大学(杭州)创新药物研究院
专家介绍:李运曼

药理学博士,教授,博士生导师,中国药科大学生理教研室副主任。曾任中国药理学会心血管药理专业委员会第十一届委员会委员,江苏省生理学会常务理事,江苏省药理学会理事,国家863计划基金评审专家,国家自然科学基金评审专家,国家发改委药品价格评审专家,国家科技奖励评审专家,中国临床药理学与治疗学审稿专家,JournalofExperimental&ClinicalCancerResearch、JournalofPharmacyandPharmacology、ActaPharmacologicaSinica以及《中国药理学通报》《中国新药杂志》《中国药科大学学报》等杂志审稿专家。从事心脑血管药理和肿瘤多药耐药逆转剂药理研究,主持和承担了1.1类创新药甲磺酸胺银内酯B片的临床研究、新型肿瘤多药耐药逆转剂HZ08与P-糖蛋白结合模式及转运机制研究等多项国家及省部级科研课题。发表SCI文章60余篇,申请专利共12项,获发明专利证书10本,获新药证书2本,获药物临床试验批件证书4本,主编《心血管药理学》《生理学实验与指导》等教材。
正文
细胞自噬对缺血性脑卒中转归中的作用研究进展
脑卒中又称脑血管意外或脑中风,是目前全球范围内致死率和致残率最高的疾病之一。脑卒中主要分为出血性脑卒中和缺血性脑卒中,其中缺血性脑卒中约占脑卒中发生率的85%。缺血性脑卒中的发生将导致神经元死亡、炎症生成和神经血管的破坏,伴随着一系列神经功能缺陷。药物治疗是缺血性脑卒中的主要治疗手段,目前唯一有效治疗脑卒中的药物是重组型纤溶酶原激活剂(recombinanttissueplasminogenactivator,rtPA),但由于治疗窗很窄而限制了rtPA的使用。因此,寻找能有效调节脑卒中的靶点迫在眉睫。
自噬是细胞利用溶酶体降解自身受损的细胞器和包括蛋白质、脂质在内的大分子物质的过程,属于非含半胱氨酸的天冬氨酸蛋白水解酶(caspase)依赖性细胞程序性死亡。细胞中存在一种将细胞内容物转运至溶酶体的囊泡,这种新型运输细胞内容物的囊泡被命名为自噬体,自噬体作为细胞内微小结构,可通过透射电镜观察,这是评价细胞是否发生自噬的特征指标。饥饿、高温、缺氧和激素刺激均能诱导自噬。自噬在维持细胞内环境的稳态中起重要作用:在细胞生长过程中,会出现一些不需要的大分子或衰老的细胞器,自噬对于这些物质的清除发挥重要作用;另外,当受到外界有害刺激时,部分细胞通过自噬降解自身内容物供其他细胞摄取利用,可以在一定程度上维持细胞内环境的稳态,抵御有害刺激。
缺血性脑卒中发生后,由于血流供应中断,大脑组织处于缺氧缺糖状态,将导致缺血区内神经元凋亡和坏死。多项研究表明,缺血性脑卒中的发生将诱导细胞自噬的发生,从而抵抗环境的变化,提示自噬在脑卒中治疗中可能是一个值得关注的靶点,而自噬在脑卒中中的调控机制尚不清楚,大脑中的神经元、胶质细胞及内皮细胞的自噬等各自特点均值得进行深入探究。
1自噬的分类
根据细胞内容物运送到溶酶体的模式的不同,将自噬分为3类:微自噬、分子伴侣介导的自噬(chaperonemediatedautophagy,CMA)和巨自噬。微自噬是一种非选择性溶酶体降解过程,指溶酶体/液泡膜内陷形成囊泡,吞噬细胞质部分。研究证实,微自噬的功能主要包括长寿命蛋白质和膜蛋白的基础降解,并且可以通过消耗多余的膜来维持和调节膜稳态与溶酶体的大小。有文献报道,在酵母细胞中的微自噬和在哺乳动物细胞中是存在差异的。酵母细胞的微自噬指的是通过液泡膜的内陷形成囊泡,从而吞噬细胞质成分;而在哺乳动物细胞中的微自噬指的是一种以热休克蛋白70(heatshockproteins,Hsc70)依赖性方式将细胞溶质可溶性蛋白质递送到晚期内体/多泡体(multivesicularbodies,MVB)中的新途径,但这种方式普遍被认为是CMA。CMA指的是细胞质内的物质依赖分子伴侣才能发生自噬,是唯一选择性降解溶酶体中可溶性胞质蛋白的自噬过程。分子伴侣介导的自噬由Hsc70和受体溶酶体相关膜蛋-2A(lysosomeassociatedmembraneprotein-2A,LAMP-2A)调节:连接着Hsc的底物复合物与LAMP-2A相互作用,将蛋白质易位至溶酶体进行消化。巨自噬指的是由一系列分子介导的双层膜结构的分解代谢过程,这种双层膜结构称为自噬体,自噬体再与溶酶体结合形成自噬溶酶体,发挥降解作用,通常所说的自噬一般就是指巨自噬。
2自噬的调控机制
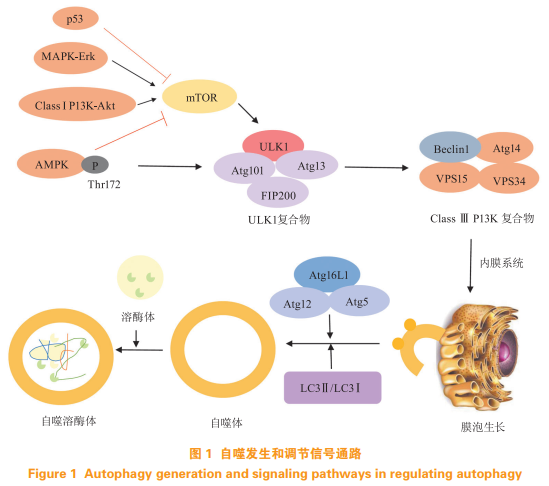
自噬的发生是通过自噬相关蛋白的协同作用进行(见图1),其中人类丝氨酸/苏氨酸蛋白激酶(humanserine/threonine-proteinkinase,又名unc-51-likekinase1,ULK1)复合物和自噬磷酸肌醇-3-激酶3(phosphatidylinositide3-kinaseclass3,PIK3C3)/液泡蛋白分选34(vacuolarproteinsorting34,VPS34)复合物是自噬体形成和自噬发生最上游和最关键调节蛋白。ULK1复合物由ULK1/2、黏着斑激酶家族相互作用蛋白200(focaladhesionkinasefamilyinteractingprotein200,FIP200)、自噬相关基因13(autophagyassociatedgene,ATG13)和ATG101组成,其中ATG13和FIP200可发挥稳定ULK1,增加其激酶活性的作用,并且在ULK1复合物从细胞质转移至内质网中发挥作用。外界刺激或上游蛋白激活ULK1复合物的方式存在争议,有文献报道认为是通过磷酸化ULK1复合物从而激活ULK1,也有文献认为是直接使ULK1复合物的表达上调来激活ULK1。ULK1的激活将直接作用于PIK3C3/VPS34复合物,后者是由苄氯素(benzylchloride,BECN1,又名Beclin-1)、ATG14、VPS15和VPS34组成,其中Beclin-1被认为是自噬发生的关键蛋白,常用来鉴定自噬是否发生。PIK3C3/VPS34复合物被激活后将与ULK1复合物一起转运至内质网上,在其他分子的协同作用下招募细胞内膜系统在内质网上形成囊泡凸起。在囊泡延长形成自噬体的过程中,2种泛素样共轭系统即ATG5-ATG12-ATG16系统和ATG4B-ATG3-ATG7-微管相关蛋白1轻链3(microtubuleassociatedprotein1lightchain3,LC3)系统起着重要作用。后者通过ATG4B将LC3切割成LC3-Ⅰ,再通过ATG3和ATG7将LC3-Ⅰ切割成LC3-Ⅱ,最后LC3-Ⅱ与磷脂酰乙醇胺(phosphatidylethanolamine,PE)结合形成完整的自噬体;而前者对于囊泡延长和LC3-Ⅱ的形成也起着非常重要的作用。自噬体形成后依靠驱动蛋白和动力蛋白沿着微管运动,最后与细胞内的溶酶体融合形成自噬溶酶体,在自噬溶酶体内,原来自噬体包裹着的生物大分子被降解。
ULK1复合物作为自噬发生最上游和最关键调节蛋白,其自身的调控和激活非常重要。研究证实,当机体能量代谢异常时,腺苷5'-磷酸依赖的蛋白激酶(adenosine5'-monophosphatekinase,AMPK)-哺乳动物雷帕霉素受体(mammaliantargetofrapamycin,mTOR)信号通路是调控ULK1表达的重要上游蛋白(见图1)。当机体能量代谢异常时,AMPK的苏氨酸172(Thr172)位点磷酸化被激活,从而抑制下游mTOR复合物,mTOR复合物的抑制将解除mTOR对ULK1的丝氨酸757(Ser757)磷酸化的抑制,使得ULK1磷酸化过度表达,自噬发生。同时,AMPK也可直接作用于ULK1,使得ULK1直接磷酸化从而激活自噬信号通路。这2条信号通路形成环路,对自噬起着调控和负调控的作用,从而维持机体内环境的稳定。
除了AMPK以外,自噬磷酸肌醇-3-激酶1(classIphosphatidylinositide3-kinase,classIPI3K)-蛋白激酶B(proteinkinaseB,又名AKT)也是调节mTOR的重要上游信号通路(见图1)。与AMPK通路是由于缺氧和能量代谢异常被激活不同,PI3KAKT信号通路一般是因生长因子作用于细胞膜上受体而被激活的,细胞膜上的受体一般指2种类型的受体,一种是酪氨酸激酶受体(receptortyrosinekinases,RTKs),另一种是G蛋白偶联受体(Gprotein-coupledreceptors,GPCRs)。在生长因子的作用下,PI3K被激活,导致AKT的Thr308磷酸化。AKT的下游是由结节性硬化复合物(tuberoussclerosiscomplex):Tsc1、Tsc2和TBC1D7组成的蛋白复合物,p-AKT可以磷酸化Tsc2并诱导其与Tsc1解离。Tsc1/2的激活使鸟嘌呤三核苷酸磷酸酶(guaninetrinucleotidephosphatase,GTP酶)Rheb失活,Rheb在GTP和GDP结合形式之间转换中起分子开关的作用。由于Tsc1与Tsc2的解离导致Rheb的失活,从而促进mTOR的磷酸化,抑制下游自噬关键蛋白ULK1的表达,导致自噬发生受到抑制。
研究证明,抑制丝裂原活化蛋白激酶(mitogenactivatedproteinkinases,MAPK)/细胞外信号调节激酶(extracellular-signal-regulatedkinase,ERK)可有效抑制自噬体的形成(见图1),说明MAPK/ERK作为mTOR上游信号分子,具有与PI3K-AKT类似的作用。p53蛋白是常见的凋亡相关蛋白(见图1),但关于p53在自噬方面的研究尚不多,最近也有文献报道p53具有抑制mTOR从而促进自噬发生的作用,尚需对p53的作用进行进一步研究。
3自噬与缺血性脑卒中
大脑中存在神经元、胶质细胞(包括星形胶质细胞、小胶质细胞)以及组成血脑屏障(blood-brainbarrier,BBB)的内皮细胞,这些细胞在大脑中的组成、所在位置和分工各有不同,在脑卒中发生后这些细胞均可以通过自噬影响疾病的转归,尽管如此,这些细胞的自噬与脑卒中的转归也存在细微的差别。
3.1神经元自噬与缺血性脑卒中
早期研究注意到缺血性脑卒中的神经元自噬,是因为神经元是缺血性损伤时最敏感的细胞。早在20世纪90年代,人们就发现大脑海马CA1区神经元在缺血性损伤后显示出含有细胞内成分的膜结合空泡,其成分来源于多种细胞,呈现无序状,后经证实这种膜结合空泡就是自噬泡。最近,利用绿色荧光标记的方式检测到在脑缺血后缺血侧脑组织神经元中自噬相关蛋白Beclin1和LC3-Ⅱ的表达,而在神经元体外培养一段时间,利用厄尔平衡盐溶液(Earle’sbalancedsaltsolution,EBSS)孵育诱导饥饿模型后也发现有绿色荧光标记的LC3-Ⅱ。
事实上,许多研究证实神经元的死亡与自噬的发生密切相关,因此就提出一种说法,即自噬是缺血性脑卒中发生后的一种细胞死亡方式,并认为自噬是有害的,抑制自噬可在一定程度上促进疾病的转归,减少神经元的损耗。Zhao等证明缺乏叶酸可增强缺血性脑卒中后神经元的自噬,从而加重脑组织损伤。还有研究证实,牡荆素可减少缺血性脑卒中后神经元的自噬从而减轻脑组织损伤,使用雷帕霉素(mTOR抑制剂,常用作自噬激动剂)后可逆转这一过程,该实验同样说明了自噬对脑卒中是一种不利因素。研究人员还发现,使用小干扰RNA(siRNA)沉默Beclin-1或Atg7可抑制神经元自噬并保护大鼠免受缺氧复氧引起的兴奋性毒性损伤。
然而,也有人提出部分神经元通过自噬降解自身内容物并形成营养成分供缺血半暗带区神经元摄取利用,以应对缺血性脑卒中发生后缺血大脑缺氧缺糖的不利环境。3-MA(一种自噬抑制剂)预处理大脑中动脉闭塞再灌注模型(middlecerebralarteryocclusion/reperfusion,MCAO/R)的啮齿类动物后,发现3-MA处理后出现有害的神经作用,使得神经元数目减少,且会导致凋亡细胞增多。有研究证明,大脑缺血再灌注后诱导自噬的发生可使神经元免受缺氧缺糖损伤,且该作用可通过清除损伤的线粒体来实现。笔者所在课题组的研究表明,使用一定浓度的AICAR(一种AMPK激动剂)在原代神经元氧糖剥夺/复氧模型(oxygen-glucosedeprivation/reoxygenation,OGD/R)孵育2h可有效提高神经元的死亡率,且使用免疫荧光法检测神经元中LC3-Ⅱ表达量升高,说明一定程度的自噬对缺血性脑卒中的恢复是有利的。笔者所在课题组研究的一种环维黄杨星D的结构类似物可展现出与AICAR类似的效果,提示该药物也可能通过诱导神经元自噬发挥对缺血性脑卒中的治疗作用。同时,大脑中烟酰胺磷酸核糖基转移酶(nicotinamidephosphoribosyltransferase,Nampt)可通过激活AMPK途径诱导自噬,并且自噬是在脑缺血早期表现出神经保护作用,这个结果更加证实细胞通过降解自身内容物来减少神经元的凋亡和坏死这个猜想,因为在脑缺血后期损伤的神经元往往无法挽救。
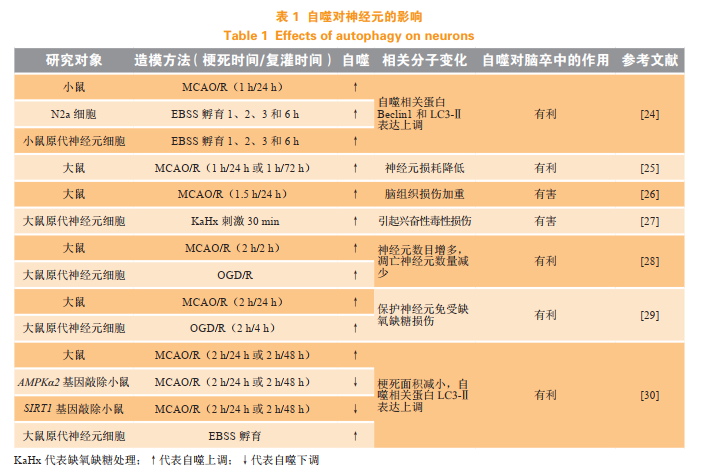
总之,自噬通过维持内环境稳态在应对外界有害刺激时起重要的作用,自噬可以保护细胞免受药物、环境毒素或疾病(包括神经退行性疾病、心血管疾病、自身免疫/炎症、代谢和年龄相关疾病)引发的各种损伤;另一方面,当自噬广泛或激活不当时,可导致细胞凋亡(程序性细胞死亡Ⅰ型),或作为替代细胞死亡途径(程序性细胞死亡Ⅱ型)发挥作用。因此自噬对于脑卒中的转归是有利还是有害,这跟疾病进程和发生自噬的细胞种类有着密切联系,不能一概而论。表1总结了近些年文献报道的神经元自噬与缺血性脑卒中的关系研究情况。
3.2胶质细胞自噬与缺血性脑卒中
胶质细胞占脑组织细胞的50%以上,在调节神经功能、维持内环境稳态等方面发挥重要作用。越来越多的证据表明,神经胶质细胞在控制神经回路、水分运输、离子稳态、免疫/炎症、神经营养因子的产生、脑血管收缩/舒张和神经发生中作用重大。
星形胶质细胞是中枢神经系统中最丰富的细胞,通过细胞与细胞之间的接触和相互作用来影响实质细胞,从而在维持正常脑功能中发挥重要作用。缺血性脑卒中发生后伴随着星形胶质细胞的激活,活化的星形胶质细胞可加剧炎症反应和神经元组织损伤,实验证实敲除circRNAHECTD1可通过抑制自噬进而抑制星形胶质细胞的活化,从而减小MCAO/R后小鼠的梗死面积,说明通过抑制胶质细胞自噬可改善缺血性脑卒中造成的脑损伤。另有文献报道,银杏内酯K可通过AMPK/mTOR/ULK1信号通路诱导星形胶质细胞自噬,使星形胶质细胞中Beclin1和LC3-Ⅱ的表达上调,从而促进氧糖剥夺后星形胶质细胞增殖和迁移,达到改善缺血性脑卒中的目的。缺血性脑卒中发生后,星形胶质细胞外钾水平显著增加,细胞内招募PI3K以激活Kir4.1(一种K+通道),导致哺乳动物雷帕霉素受体复合物1(mammaliantargetofrapamycincomplex1,mTORC1)和整个mTOR途径被激活,从而诱导自噬产生,致使缺血半暗带中更多细胞存活且脑损伤减少。但该实验提到,Kir4.1诱导自噬保护脑组织的作用也具有时间特异性。
小胶质细胞激活和M1小胶质细胞/巨噬细胞极化的促进在脑神经炎症中起重要作用。M2小胶质细胞通过产生白细胞介素4(interleukin4,IL-4)和IL-10等抗炎因子去除细胞碎片和保护相邻细胞。实验证实,自噬也会作用于小胶质细胞/巨噬细胞极化的炎症反应中:miR-30d-5p过表达的脂肪干细胞产生外泌体,可有效抑制OGD/R造模后原代小胶质细胞自噬,从而促进小胶质细胞的M2型极化,减轻脑组织损伤。实验表明,使用3-MA(一种自噬抑制剂)后可减少小胶质细胞的自噬和炎症的发生,并可有效减少梗死面积、脑水肿和神经功能评分,提示缺血性脑卒中诱导的小胶质细胞自噬会加重缺血性神经炎症和损伤。有研究还发现,四环素可通过抑制小胶质细胞的自噬来抑制神经炎症,从而改善脑组织的损伤。尽管以上研究证实小胶质细胞自噬会加重缺血性脑卒中造成的脑组织损伤,但由于相对研究较少,实验数据不够充分,因此小胶质细胞自噬与脑卒中的关系尚需进一步探究。
3.3内皮细胞自噬与缺血性脑卒中
BBB是指位于血液与脑、脊髓的神经细胞之间物质交换限制系统,由毛细血管紧密连接的内皮细胞层、基膜和星形胶质细胞足突组成。缺血性脑卒中发生后,BBB的完整性受损,其对物质的选择性渗透也遭到破坏,因此,缺血性脑卒中发生后,组成BBB的内皮细胞的功能恢复显得尤为重要。
大多数BBB被破坏的研究均集中于基质金属蛋白酶(matrixmetalloproteinase,MMP)介导的紧密连接蛋白降解,但最近几项研究表明自噬也可能起重要作用。神经突起导向因子-1(Netrin-1)输注可改善MCAO/R模型大鼠神经功能,减少BBB继发性损伤,降低蛋白质外渗,并且在内皮细胞体外实验中发现该作用在使用3-MA后消失,说明Netrin-1可能是通过诱导内皮细胞自噬发挥对脑组织的保护作用。研究人员还发现,非编码RNAMalat1对脑卒中发生后的大脑有保护作用,可诱导脑微血管内皮细胞LC3-Ⅱ和p62表达上调,体内外研究证实非编码RNAMalat1是一种有效的自噬诱导剂,通过刺激miR-26b和上调ULK2表达诱导自噬发生,保护脑微血管内皮细胞免受OGD/R的损伤。甲基乙二醛(MGO)是葡萄糖代谢过程中产生的反应性二羰基化合物,可加剧缺血诱导的内皮细胞损伤和功能障碍。研究发现,自噬对MGO诱导的内皮细胞损伤具有保护作用,可减小MCAO/R后大脑的梗死面积,减轻BBB的损伤;使用3-MA则会加重MGO引起的内皮细胞损伤,这也提示内皮细胞的自噬具有保护脑组织的作用。
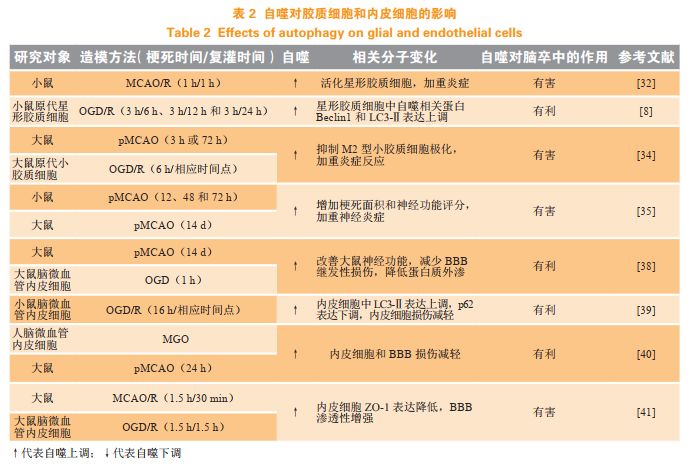
但体外给予脑微血管内皮细胞雷帕霉素将诱导自噬,同时造成紧密连接蛋白ZO-1表达降低,BBB渗透性增强,BBB的功能受损,提示自噬的发生也有可能会加重BBB的损伤,不利于脑卒中的转归。总之,尽管对于内皮细胞自噬的研究已有很多,但内皮细胞自噬在缺血性脑卒中中的双向作用仍值得进行深入研究。表2总结了近些年文献报道的胶质细胞和内皮细胞自噬与缺血性脑卒中的关系研究情况。
4结语与展望
缺血性脑卒中发生后,由于缺氧缺糖导致脑细胞,尤其是神经元的损伤是缺血性脑卒中发生后神经功能缺陷的主要原因,在缺血核心区的神经元坏死或凋亡无法挽救,而在缺血核心区周围存在由于供血不足产生的缺血半暗带,半暗带中的细胞由于缺乏营养物质岌岌可危。自噬是一把双刃剑,适当的自噬能够降解细胞自身内容物供周围细胞吸收利用,从而挽救由于缺血濒临死亡的细胞;另一方面,过度的自噬将导致细胞大量死亡,对于缺血性脑卒中来说,与细胞凋亡或坏死没有本质区别,将加重缺血性脑卒中引起的脑组织损伤。脑组织中有大量的神经元、包括星形胶质细胞和小胶质细胞在内的胶质细胞以及组成BBB的内皮细胞,这些细胞在调节大脑神经功能,维持大脑内环境稳态中起重要作用,因此除了研究自噬的利与弊外,研究大脑中不同的细胞自噬对寻找缺血性脑卒中治疗新靶点具有重要意义。
迄今为止,脑卒中依然是世界上发病率和致死率最高的疾病之一,因此,寻找治疗缺血性脑卒中的靶点和药物非常重要。自噬作为机体维持稳态的一种程序性死亡途径,是调节缺血性脑卒中发生后大脑中细胞凋亡、坏死和炎症发生的关键,故将自噬作为开发缺血性脑卒中治疗药物的靶点是具有现实意义的。由于目前的研究尚未对自噬与缺血性脑卒中的关系有深刻且明确的认识,因此,未来可从自噬发生的时间、自噬调节的程度等方面着手研究自噬与缺血性脑卒中的关系,这对以自噬作为靶点的缺血性脑卒中治疗药物的开发具有重要意义。
来源:ppsyxjz 药学进展
原文链接:http://mp.weixin.qq.com/s?__biz=MzA5MDY3ODExNQ==&mid=2651307457&idx=1&sn=fc62dbe7c26d7fb9c10448e27e23ad17&chksm=8bf4ebcdbc8362db6d498d46552235df3e60ceda1c41eabb4db4963fa4006cea4013fa55abc5&scene=27#wechat_redirect
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
小胶质细胞 脑组织 内皮细胞 神经元细胞 溶酶体 细胞自噬 缺血性脑卒中
神经元释放的IL-33介导小胶质细胞吞噬细胞外基质促进突触重塑
小胶质细胞监测和保护神经元的功能

科学家实现将人类干细胞转化为类脑组织的尖端技术
生活急救-脑卒中-2(缺血性脑卒中-2)
Ucp2依赖的小胶质细胞-神经元耦合调控焦虑样行为
简单小分子组合实现胶质细胞直接转化为神经元

中文解读 | CCM3在颅内海绵状血管畸形中的作用
《自然》:小胶质细胞或能有效调节大脑中神经元的功能和行为!

学科动态 ▏护理团体标准解读:缺血性脑卒中静脉溶栓护理
Treg细胞调节缺血性脑卒中后免疫反应


科界APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号