科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-11-28
来源:BioArt
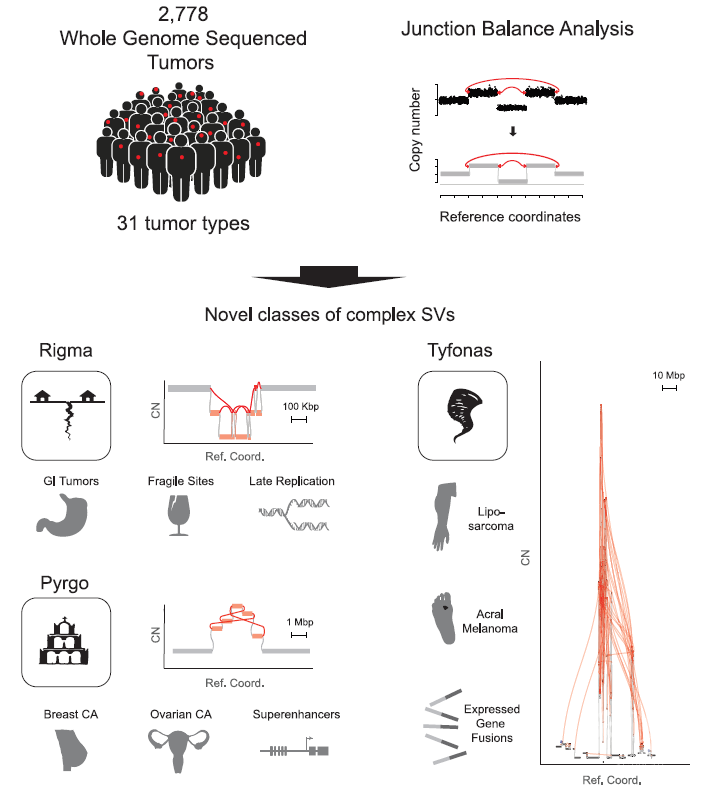
癌症基因组在多种突变机制的作用下,会产生丰富的结构变异(structural variation)。简单的结构变异模式如删除(deletion)、串联重复(tandem duplication)只由一个或两个位点断裂-融合组成新的链接(junction),而复杂的结构变异则会引起多个链接且组合成各种复杂的序列,通常还会伴随拷贝数变异(copy number aberration),例如染色体碎裂(chromothripsis)、复杂染色体重排(chromoplexy)、断裂-融合-桥染色体循环(breakage-fusion-bridge cycle)。虽然通过全基因组测序(whole genome sequencing),人们已经可以识别变异后的DNA链接和拷贝数变异,但仍没有一套统一的方法来建立重排后的DNA模型用来发掘所有的变异模式。
近日,康奈尔医学院和纽约基因组中心的Marcin Imielinski实验室在Cell上发表了文章Distinct Classes of Complex Structural Variation Uncovered across Thousands of CancerGenomeGraphs,基于一个关于DNA的简单事实设计了链接平衡算法(Junction Balance Analysis,JaBbA),来把癌症基因组中的链接和拷贝数统一起来,重建出量化的基因组图(genome graph),用于识别任何复杂度的结构变异。由于DNA的线性化学结构,除末端外每个片段都只且必有一个上游的相邻片段和一个下游的相邻片段,即每个独特片段的总拷贝数必须与上游的的链接拷贝数之和、下游的的链接拷贝数之和相等。在此限制条件下,通过混合整数规划,得到全基因组尺度上的最优整数解,用一个统一的模型同时推断了DNA片段以及相邻DNA片段之间的链接拷贝数(junction copy number)。在全面的基准测试证明JaBbA的DNA片段和链接拷贝数的准确性后,作者在2778个泛癌种全基因组数据上重建了基因组图模型,并从低(1或2)和高拷贝数链接(大于7)两个极端对链接簇的组成模式进行挖掘。

作者首先发现,在全部由低拷贝链接组成的链接簇中,富集了两大类别,其一几乎全部是删除链接组成,二是几乎全部由串联重复组成。用伽马-泊松回归估计每一个基因组中每个固定长度区段上的删除链接的分布后,得到一系列过量富集删除链接的区段。这样的区段中多个删除链接作用于同一位点,导致拷贝数断崖式下降,象形取名为基因组断层(rigma)。基因组断层在食道腺癌、巴雷特食道症(Barrett's esophagus)、胃癌、大肠癌等多种消化道病变中最为常见,在较晚复制的基因组区域中富集,并且特别集中于长基因(大于1兆碱基对)中。有趣的是,最常重复出现基因组断层的区域与已知的人类基因组的脆性位点(fragile site)高度重合,包括FHIT、MACROD2、LSAMP等基因,但也包含许多还未被标记为脆性位点的区域。由于已知的脆性位点大多是通过细胞实验或胚系突变模式推断的,所以只局限在最突出的位置上,而癌症的高突变率则可能给我们指向了更多还未被发现的脆性位点。
用同样的方法观察串联重复链接,作者发现广泛存在的多个串联重复链接簇,在同一位点上多次复制,状似金字塔,故名为基因组塔(pyrgo)。这类结构变异在子宫内膜癌、卵巢癌、乳腺癌和食道腺癌中最常出现,且过量富集于超增强子(superenhancer)所在的区域。与基因组断层相反,基因组塔更偏好较早复制的区域。MYC基因位点是基因组塔最频繁发生的位置之一,而其中MYC上游的超级增强子比之基因本身更经常被扩增,作者认为这与串联重复调控元件来刺激靶基因的假说不谋而合。
另一方面,在高拷贝数即扩增的区域中,作者对所有含有高拷贝数链接的链接簇根据三个特征进行了聚类分析,除了发现了已知的二重微小染色体(double minute)和断裂-融合-桥循环两种模式之外,还观察到了一类新的具有大量高拷贝链接、大量折返式链接(fold-back)的扩增模式。其影响范围可多达数个染色体,被扩增的DNA总量能达到100~200百万碱基对,命名为基因组台风(tyfonas)。这种模式在近50%的肢端黑色素瘤和近80%的脂肪肉瘤中存在,而在其他类型的黑色素瘤与软组织肉瘤中则非常罕见。与TCGA样本中通过RNA-seq发现的融合基因转录本比对,发现基因组台风比其他复杂突变能更高效的产生融合基因,这也和肢端黑色素瘤患者通常缺乏小突变新抗原却对免疫检查点抑制剂敏感产生了联系。另外,结构变异链接端附近的单碱基突变频率也局部升高,在基因组台风的链接周围尤其明显。结合这些观察,作者进一步通过染色质构象和光学位点比对的数据提出了一种可能造成基因组台风的机制,即早期的染色体碎裂形成的短片段被随机修复并进行了线性和环状的折返式扩增,最终通过与其他染色体的结合重新获得了端粒与中心粒并稳定存在于癌细胞中。
把共13中从简单到复杂的结构变异模式整合,对全部的病人进行聚类,总结出了14大类结构变异模式的组和,每一类富集的肿瘤类型和既往观测到的现象相吻合。比如前列腺癌在碎裂组和复杂染色体重排组富集,遗传性BRCA1缺陷的乳腺癌、卵巢癌在串联重复最多的组合中富集(DDT)。与缺乏结构变异的大类(QUIET)相比,有六类病人的总生存期显著缩短,标志着结构变异的归类在临床预后中的应用潜力。
作者们表示,这项研究成果证明基因组图数据结构是分析肿瘤结构变异的强有力的工具,随着数据量进一步增大还会有更多的突变模式被发现,并与背后的的病因联系更紧密,最终进一步推动全基因组测序走向临床应用。
专家点评
叶凯(西安交通大学)
基因组变异包括单碱基替换变异(SNV)、短插入缺失变异(indel)和结构变异。短插入缺失变异通常定义为小于50个剪辑对的简单类型插入或者缺失变异,而广义结构变异指SNV和indel以外的所有变异。在基因组二代测序发展的早期,只有读长较短的单端测序数据,我们只能通过分析测序数据在基因组上覆盖度的差异,即read-depth方法来理解拷贝数变异;随着双端测序技术发展,我们开发出了基于测序数据两端在参考基因组上比对距离和方向的read-pair方法,发现基因组中广泛存在的各种类型结构变异。当前,世界各国高质量的二代甚至是三代测序数据大量涌现,进一步揭示了结构变异在种群差异、驱动物种性状演化、疾病发生发展中的重要作用。最近人群队列和肿瘤基因组研究揭示了复杂结构变异的存在,学界仍然在探索如何更好的发现、表征复杂类型结构变异,统一复杂结构变异的数学描述。
本文作者提出了JaBaA(Junction Balance Analysis)的分析方法,基于参考基因组以及全基因组测序数据利用初步获取片段(segmentation)的测序深度(read depth)信息以及链接(junction)信息,以片段作为节点,链接作为边构建JBGG(junction-balanced genome graph)基因组图模型。首先,假设片段内小区间(bins)的拷贝数符合高斯分布建立似然函数。然后考虑到图模型中部分loose end的边可能受低比对率、测序深度或者纯度等因素影响,对loose end节点的拷贝数做出假设,建立先验分布。作者基于贝叶斯理论推导将片段内信息(每个节点拷贝数分布的残差)与片段间的连接信息(先验中的loose end节点的出度以及入度)共同作为目标函数进行最小化,在此过程中,作者巧妙的基于一个简单的事实作为约束,即图中每个节点的拷贝数应该与它们的入度以及出度数量一致,将目标函数中两部分联系起来,使得它们可以共同作用得到最优的估计拷贝数(junction copy number)。作者在全面的基准测试证明了估计拷贝数的准确性,并在2778个泛癌症全基因组数据上重建图模型进行模式挖掘,定义了pyrgo,rigma和tyfonas等全新的复杂重排现象。由此证明了该方法在分析肿瘤结构变异中的重大作用,随着未来数据量的增加会进一步推动测序技术向临床应用的转变。
本文创新性地搭建基于基因组图数据结构的肿瘤复杂结构变异解析方法,将基于二代测序数据的变异检测提升到一个新的高度,随着该方法在大量肿瘤测序数据广泛运用,我们将发现更多以前未知的复杂结构变异,探索其发生机理、变异率、位点偏好性等表征,揭示新的肿瘤发生发展机理,为肿瘤早诊、个体化治疗方案、伴随诊断等医疗应用提供关键技术支撑,面向人民生命健康。
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652511736&idx=4&sn=e0c0bc81e82bda4202339419bee2b2d3
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn

迄今最综合癌症基因组图出炉
【病毒变异专题】SARS-CoV-2突变的结构基础

临床癌症基因组学 |《自然-癌症》专题
全基因组泛癌症分析:揭开未知的99%


单细胞基因组学推动癌症免疫疗法的研究

Nature连发6篇文章,聚焦癌症基因组学
【十万个为什么】你知道吗肿瘤的历史比人类历史还长!

Science:癌症基因组中的突变“热点”不一定会推动癌症的生长!
Nat Genet:揭秘线粒体基因组奥秘 或有望帮助开发多种癌症新型疗法

揭开未知的99%:最大规模癌症基因组研究公布


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号