科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2021-09-17
正文
BIR2 选择性凋亡抑制蛋白抑制剂研究进展
[摘要]凋亡抑制蛋白 XIAP、c-IAP1 和 c-IAP2 是抗肿瘤新药研发的重要靶点,其均含有 3 个 BIR 结构域,许多靶向 BIR2 和 BIR3结构域的小分子 IAPs 抑制剂已被报道,其中 BIR2 选择性 IAPs 抑制剂研究相对较少。不同于 BIR3 选择性 IAPs 抑制剂,BIR2 选择性IAPs 抑制剂可以阻断 XIAP 与 Caspase-3 和 Caspase-7 的相互作用,因此是一类具有不同作用机制的新型 IAPs 抑制剂,对这类化合物的研究对进一步阐明 IAPs 及 IAPs 抑制剂的作用机制具有重要意义。综述凋亡抑制蛋白的结构与功能、Smac 的功能及其与 IAPs的相互作用、BIR2 选择性 IAPs 抑制剂的研究进展,以期为相关研究提供参考。
1 凋亡抑制蛋白的结构与功能
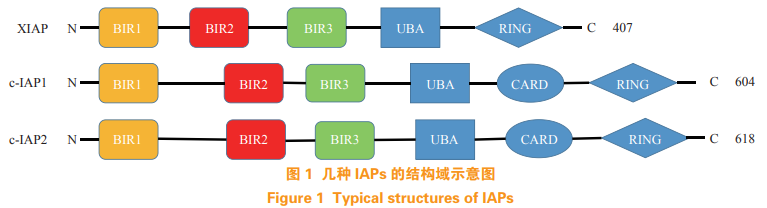
凋亡抑制蛋白(inhibitor of apoptosis proteins,IAPs)是一类高度保守的内源性抑制细胞凋亡的蛋白家族,在昆虫、酵母和哺乳动物等中广泛存在 [1]。目前在人体中发现的 IAPs 共有 8 种,包括 NIAP、c-IAP1、c-IAP2、XIAP、Survivin、BRUCE、MLIAP 和 ILP2 [2]。IAPs 蛋白家族的结构特征是氮端含有 1 ~ 3 个包含 70 个氨基酸的杆状病毒 IAP 重复序列(baculoviral IAP repeat,BIR),即 BIR 结构域。BIR 结构域是绝大多数 IAPs 的抗凋亡活性所必须的,不同 BIR 结构域具有一定的同源性,但功能却各不相同 [3]。IAPs 主要通过直接或间接地抑制半胱天冬酶(Caspases)的活性或参与调节核因子 κB(NF-κB)的功能来抑制细胞凋亡,抑制 IAPs 可以诱导细胞凋亡或增加细胞死亡的阈值 [4]。在 IAPs 中被研究最多的是 XIAP、c-IAP1 和 c-IAP2,它们均含有 3 个 BIR 结构域(见图 1)。XIAP 的 BIR3 结构域可以结合并抑制 Caspase-9,而 XIAP 的 BIR2结构域和 BIR1 与 BIR2 之间的肽链可以结合并抑制 Caspase-3 和 Caspase-7 [5]。c-IAP1 和 c-IAP2 也可以通过它们的 BIR3 结构域与 Caspase-9 结合,但不能抑制 Caspase-9 的活性,它们主要通过抑制 Caspase-8 的激活来抑制细胞凋亡的发生 [6-8]。XIAP、c-IAP1 和 c-IAP2 在很多肿瘤中过度表达,而且其过度表达与肿瘤的发展和抗药性的产生密切相关,因此它们被认为是研发新型抗肿瘤药物的靶点 [9-12]。
2 Smac 的功能及其与 IAPs 的相互作用
Smac 是线粒体接收到凋亡信号后释放出的一种蛋白,它是细胞中 IAPs 的内在调节剂。Smac 可以通过其氮端的四肽 AVPI(Ala-Val-Pro-Ile)与 IAPs的 BIR 结构域结合,因此 Smac 氮端的四肽 AVPI被称为 IAP 结合单元(IAP binding motif,IBM)。当 Smac 与 XIAP 结合时先形成二聚体,二聚体中的 2 个 AVPI 四肽单元分别与 XIAP 的 BIR2 和 BIR3结合。Smac 与 XIAP 的结合可以直接抑制 XIAP 与Caspase-3、Caspase-7、Caspase-9 的结合,从而恢复Caspase-3、Caspase-7、Caspase-9的功能,促使凋亡发生。当 Smac 与 c-IAP1 和 c-IAP2 结合时只与这 2 种 IAPs的 BIR3 结合。Smac 与 c-IAP1 和 c-IAP2 结合后会诱导它们降解,从而间接促使 Caspase-8 的激活 [9-12]。
目前已有多个以 XIAP、c-IAP1 和 c-IAP2 为靶点的小分子 IAPs 抑制剂进入临床研究,其中大部分都是以四肽 AVPI 为先导化合物设计的 BIR3 选择性泛 IAPs 抑制剂,即这些化合物可以与 XIAP、c-IAP1 和 c-IAP2 的 BIR3 结构域有效结合 [13]。另外,基于 BIR3 选择性 IAPs 抑制剂已有很多报道,其中部分化合物已经进入临床研究 [13-14]。对 BIR2选择性 IAPs 抑制剂的研究相对较少,但近年来随着对 IAPs 作用机制的深入研究,对 BIR2 选择性 IAPs抑制剂的研究也引起了越来越多药物化学家们的关注。BIR2 选择性 IAPs 抑制剂可以阻断 XIAP 与Caspase-3 和 Caspase-7 的相互作用,但不会使 c-IAP1和 c-IAP2 降 解, 也 不 会 引 起 因 c-IAP1 和 c-IAP2降解导致的非经典 NF-κB 通路的激活,其功能与BIR3 选择性 IAPs 抑制剂完全不同,因此是一类新型的 IAPs 抑制剂,不仅可以作为药物进行研发,也可以作为小分子工具用于相关的生物学机制研究。
3 BIR2 选择性 IAPs 抑制剂
3.1 早期通过高通量筛选得到的 BIR2 选择性 IAPs 抑制剂
对 BIR2 选择性的 XIAP 抑制剂的研究最早可追溯至 2003 年。Tamm 等 [15] 利用噬菌体展示技术发现一个通过二硫键环合的六肽 CEFESC(Cys-Glu-PheGlu-Ser-Cys)可以特异性地与 XIAP 的 BIR2 结构域相结合,但跟其他 IAPs 没有结合作用。Caspase-3和 Caspase-7 可抑制该六肽 CEFESC 与 XIAP 的结合,但 Caspase-8 不具备该功能,表明该六肽是靶向XIAP 中与 Caspase-3 和 Caspase-7 结合的区域,但该六肽与 XIAP 的具体结合模式尚不清楚。
2003 年 Wu 等 [16] 通过高通量筛选的方法对一个包含 160 000 个小分子化合物的化合物库进行了筛选,成功发现了一系列非肽类的小分子磺酰胺类化合物,这类化合物通过与 XIAP 中的 linker-BIR2 区域结合,竞争性地解除 XIAP 对 Caspase-3 的抑制作用,其中化合物 1 结合的 IC50 为 10 µmol·L-1。这类化合物可以诱导 293 细胞系的凋亡,并且增强 CD95和肿瘤坏死因子相关的凋亡诱导配体(TNF relatedapoptosis inducing ligand,TRAIL)诱导凋亡的能力。
2004 年 Shimmer 等 [17] 通过对包含近 1×108 个肽类及非肽类化合物的化合物库进行高通量筛选,发现了一系列多苯基脲类BIR2选择性XIAP抑制剂,其中活性最强的是化合物 2。该化合物可以特异性与 XIAP 的 BIR2 结构域结合并恢复 Caspase-3 的活性,而且可以直接诱导多种肿瘤细胞凋亡。在异种移植的小鼠模型中该化合物可以抑制肿瘤组织的生长并增加肿瘤对化疗药物的敏感性。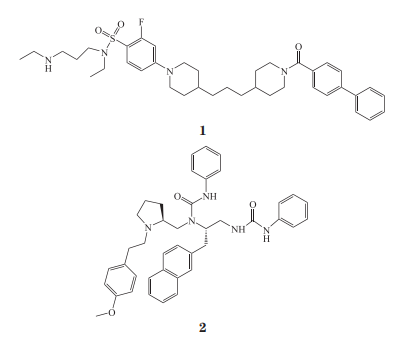
3.2 以多肽为先导化合物设计的 BIR2 选择性 IAPs 抑制剂
虽然以往研究中通过高通量筛选发现了一些 BIR2 选择性 XIAP 抑制剂,但这些化合物与XIAP-BIR2 的结合能力通常不强,而且结合模式也不确定,因此多个课题组采用与 BIR3 选择性 IAPs抑制剂的研发中类似的方式,以 Smac 氮端的四肽AVPI 为先导化合物,对 BIR2 选择性 IAPs 抑制剂进行研究。
四肽 AVPI 既可以与 XIAP 的 BIR2 结构域结合,也可以与 XIAP 的 BIR3 结构域结合,但它与BIR3 的结合能力强于与 BIR2 的结合能力。2006 年Sweeney 等 [18] 用不同的天然氨基酸对 AVPI 中各个位置的氨基酸分别进行替换,合成了一系列四肽,并研究了四肽与 XIAP 的 BIR2 结构域结合的构效关系。结果表明,为了保持与 XIAP-BIR2 的结合能力,P1 位置只能选 Ala;P2 位置倾向于 β 位带有支链的疏水性氨基酸,如 Val、Thr、Ile 等,或者是既含有疏水性,又含有亲水性片段的氨基酸,如 Gln、Glu、Lys、Arg、Tyr 等;P3 位置倾向于含有小的侧链的氨基酸,如 Ala、Ser、Val 等;P4 位置也倾向于带有较小侧链的氨基酸,如 Gly、Ala、Thr、Val 等。
2013 年 Lukacs 等 [19] 对 Sweeney 等 [18] 的研究结果进行了确证,发现与 BIR2 结合能力最强且选择性最高的四肽是 ATAA(Ala-Thr-Ala-Ala),该四肽ATAA与XIAP的BIR2结合的抑制常数Ki为1.7µmol· L-1, 比 Smac 氮端的四肽 AVPI 与 XIAP 的BIR2 的结合能力强 3 倍(Ki = 5.2 µmol·L-1)。更重要的是,Lukacs 等 [19] 还解析了 5 个四肽与 XIAP 的BIR2 形成的复合物的晶体结构,分别是 ATAA 复合物(PDB ID:4j45)、AIAV 复合物(PDB ID:4j44)、AVPI 复合物(PDB ID:4j46)、SVPI 复合 物(PDB ID:4j47) 和 AMRV 复 合 物(PDBID:4j48),从而从结构生物学的角度证实了这些四肽与 XIAP 的 BIR2 结构域的结合模式。虽然这些四肽与 XIAP-BIR2 的结合能力有差异,但它们与 XIAP-BIR2 的结合模式基本一致。比较 AVPI 与XIAP-BIR2 和 XIAP-BIR3 形成的复合物的晶体结构可以发现,BIR2 和 BIR3 中 3 个关键位置的氨基酸残基的不同显著影响了它们与四肽的结合,这 3 个位置的氨基酸残基分别是 His223/Trp323、Phe224/Tyr324 和 Lys206+Lys208/Gly306+Thr308(见图 2)。四肽中 P3 位置的氨基酸残基可以与 His223/Trp323和 Phe224/Tyr324 结合。His223 中的侧链比 Trp323中的侧链小,因此 BIR2 与四肽中 P3 位置的氨基酸残基的疏水性作用弱于 BIR3。同样因为 His223 中的侧链小于 Trp323,His323 与 Phe224 之间的空腔比 Trp223 与 Tyr324 间的空腔大,而且 Tyr324 中的羟基可以与 BIR3 中的 Gly306 的羰基形成氢键,使蛋白构象具有更强的刚性,所以与 BIR2 结合时四肽的 P3 位置可以是较大的不同基团,而与 BIR3 结合时 P3 位置只能是 Pro。在 XIAP-BIR2 中 Lys206和 Lys208 的侧链及其下方的 Gln197 的侧链形成了一个较浅的口袋与四肽中 P4 位置的氨基酸残基的侧链结合,而在 XIAP-BIR3 中对应于 Lys206 和Lys208 的分别是 Gly306 和 Thr308,这 2 个残基与P4 位置的氨基酸侧链结合较弱,P4 位置的氨基酸侧链主要是与其下方由 Lys297、Lys299 及 Leu292 形成的一个较大的疏水性口袋结合,因此 XIAP-BIR3中与 P4 位置结合的疏水性空腔较大,可以容纳较大的疏水性氨基酸侧链。这些结构信息为设计选择性IAPs 抑制剂奠定了基础。
2011 年 Cosford 课题组发现将 AVPI 四肽中的Ile 残基用芳基取代可以提高化合物对 BIR2 的结合能力,其中选择性最高的化合物 3 与 XIAP 的 BIR2结合的 Ki 为 0.74 µmol· L-1,是该化合物与 XIAP的 BIR3 结合能力的 7 倍(Ki = 5.5 µmol· L-1)[20]。Cosford 课题组在这类化合物中用氮甲基丙氨酸替换了 AVPI 中的 Ala,因为以前在 BIR3 选择性的化合物的研究中发现在 Ala 的氨基上引入甲基不会影响化合物的蛋白结合能力,但可以显著提高化合物的抑制肿瘤细胞生长的能力,可能是因为引入甲基后可以提高细胞穿透性。细胞活性研究表明,这类化合物在乳腺癌 MDA-MB-231 细胞系中可以增强TRAIL 引起凋亡的能力。在上述研究的基础上,Cosford 课题组对该类化合物的构效关系进行了进一步的探索,发现用 Ala 替换 Pro 可以进一步地提高选择性,其中选择性最优的化合物 4 与 XIAP 的BIR2 的结合能力是其与 XIAP 的 BIR3 的结合能力的 70 倍(Ki:0.64 µmol· L-1 vs 40.15 µmol· L-1);与化合物 3 类似,化合物 4 单一给药并未显示出抑制肿瘤细胞生长的能力,但同样能使细胞对 TRAIL诱导的凋亡更加敏感 [21]。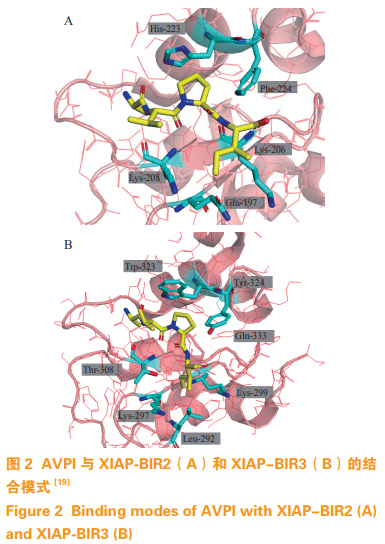
2014 年罗氏公司公开了 2 项拟肽类的 BIR2 选择性 IAPs 抑制剂的专利,发现用芳环与 AVPI 衍生物中Pro 的五元环并环后可以显著提高化合物与 BIR2 的结合能力和选择性,其中代表性的化合物 5 与 XIAPBIR2 和 XIAP-BIR3 结合的 IC50 分别为 0.013 μmol·L-1 和大于 55 μmol·L-1,化合物 6 与 XIAP- BIR2 和XIAP-BIR3 结合的 IC50 分别为 0.010 μmol·L-1 和大于55 μmol·L-1 [22-23]。这些结果进一步证明了以四肽为先导化合物研发 BIR2 选择性 IAPs 抑制剂的可行性。
3.3 经过高通量筛选并优化得到的新型 BIR2 选择性 IAPs 抑制剂
2013 年 Andrew 课题组报道了一类新型的苯并西平类和苯并二氮杂䓬类 BIR2 选择性 IAPs 抑制剂 [24-25]。该课题组首先通过高通量筛选发现了一个具有一定的 BIR2 选择性且相对分子质量与亲脂性均较低的化合物 7,随后以化合物 7 为先导化合物进行了详细的结构优化,合成了一系列 BIR2选择性小分子 IAPs 抑制剂,显著提高了化合物的蛋白结合能力和选择性,并改善了药代动力学性质,其中代表性的化合物 8 与 XIAP-BIR2 结合的IC50 为 39 nmol· L-1, 与 XIAP-BIR3 结合的 IC50 为3.06 μmol· L-1。与其选择性结合并抑制 XIAP-BIR2的活性一致,化合物 8 在体内和体外均可以有效阻断 XIAP 与 Caspase-3 和 Caspase-7 的 相 互 作 用, 而且不会引起 c-IAP1/2 的降解,表明化合物 8 并不能与 c-IAP1/2 的 BIR3 结构域有效结合。随后的研究发现,化合物 8 对细胞色素 P4503A4 酶(CYP3A4)具有很强的抑制作用(IC50=1.8 µmol·L-1),因此不能用于进一步的临床研究。为了进一步改善成药性,该课题组在化合物 8 的基础上进行了进一步广泛且细致的优化,在保持与 XIAP-BIR2 的结合能力及选择性的同时,进一步改善了化合物的药代动力学性质,并显著降低了化合物对 CYP3A4 的抑制,其中最优的化合物 9 抑制 XIAP-BIR2 和 XIAP-BIR3 的 Ki 分别为45 nmol·L-1 和 30.8 µmol·L-1。化合物 9 抑制 CYP3A4的 IC50 是 16 µmol·L-1,与化合物 8 相比显著降低。药代动力学研究表明,化合物 9 具有良好的药代动力学性质和安全性,在体内实验中化合物 9 与 DR5 抗体 conatumummab 联合用药时可以有效增强 Caspase-3和 Caspase-7 的活性。另外在机制研究中发现,化合物 9 在作用过程中没有激活非经典的 NF-κB 通路,表明该化合物不会有效抑制 c-IAP1/2 的活性。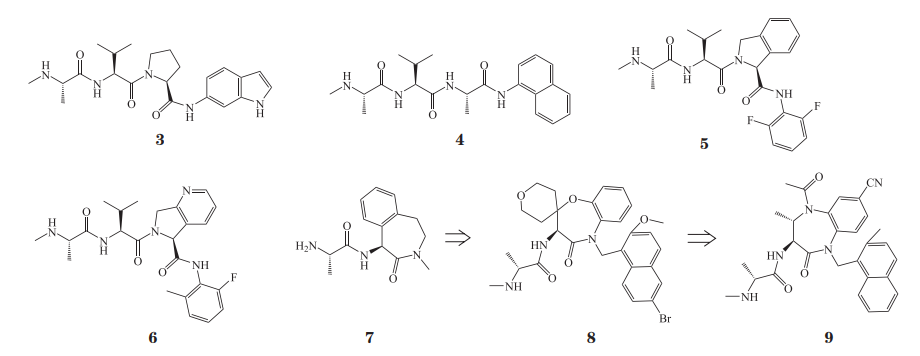
4 结语与展望
细胞凋亡是癌症的主要特征之一,在细胞凋亡中有重要功能的蛋白很多都是抗肿瘤新药研发的重要靶点。IAPs 是细胞凋亡中的逆向调节剂,已有多个靶向 IAPs 的 BIR3 结构域的小分子化合物和基于 BIR3 选择性 IAPs 抑制剂设计的二聚体 IAPs 抑制剂进入临床研究。不同于广受关注的 BIR3 选择性 IAPs 抑制剂,BIR2 选择性 IAPs 抑制剂有其独特的功能。BIR2 选择性 IAPs 抑制剂可以阻断 XIAP与 Caspase-3 和 Caspase-7 的相互作用,并促进多种抗肿瘤药物的抗肿瘤效果,但不会引起 c-IAP1 和c-IAP2 的降解,因此对 BIR2 选择性 IAPs 抑制剂的作用机制进行研究,可以进一步阐明 XIAP、c-IAP1和 c-IAP2 的生物学功能,并促进以 IAPs 为靶点的新药研发。笔者所在课题组近年来一直在从事新型IAPs 抑制剂的研发工作,近期工作集中于 BIR2 选择性 IAPs 抑制剂的设计、合成及生物活性研究,并探索 BIR2 选择性 IAPs 抑制剂的进一步利用。
在已报道的 IAPs 抑制剂中,最初研发二聚体IAPs 抑制剂的目的是发展出能够模拟二聚的 Smac蛋白的功能,同时与 XIAP 的 BIR2 和 BIR3 更有效结合的 XIAP 抑制剂,但已报道的二聚体 IAPs 抑制剂实际上都是基于 BIR3 选择性 IAPs 抑制剂设计的,利用 BIR2 选择性的化合物与 BIR3 选择性的化合物有可能设计出更有效的二聚体 XIAP 抑制剂。另外,已报道的二聚体 IAPs 抑制剂的抗肿瘤活性比 BIR3选择性的单体 IAPs 抑制剂强 100 倍以上,而它们与 XIAP 的结合能力也比单体 IAPs 抑制剂强 100 倍以上;但有研究表明,IAPs 抑制剂的抗肿瘤活性并不依赖于对 XIAP 的抑制,对其具体作用机制尚不清楚。因此,通过设计 BIR2 选择性 IAPs 抑制剂和BIR3 选择性 IAPs 抑制剂组成的新型二聚体 IAPs 抑制剂,有可能进一步阐明二聚体 IAPs 抑制剂的作用机制,而且可以促进二聚体 IAPs 抑制剂在新药研发中的应用。
近年来,对蛋白降解靶向嵌合体(PROTACs)的 研 究 已 成 为 药 物 化 学 研 究 中 的 热 点 之 一。PROTACs 是由与靶蛋白结合的配体和与 E3 泛素连接酶结合的配体通过连接链连接而成的一类嵌合分子,可以同时与靶蛋白和 E3 泛素连接酶结合,促进靶蛋白的泛素化,使靶蛋白被蛋白酶体降解。XIAP、c-IAP1 和 c-IAP2 均具有 E3 泛素连接酶的功能,因此 IAPs 抑制剂可被用于 PROTACs 的研发,但 BIR3 选择性 IAPs 抑制剂可以诱导 c-IAP1和 c-IAP2 的快速降解,因此严重限制了它们在PROTACs 研发中的利用。BIR2 选择性 IAPs 抑制剂不会引起 c-IAP1 和 c-IAP2 的降解,因此有可能在基于 IAPs 的 PROTACs 的研发中得到应用。
最后,选择性地作用于 cIAP1 和 cIAP2 的小分子化合物已有报道,但选择性地作用于 XIAP 的小分子化合物一直未能发现。以前的研究结果表明,BIR2选择性 IAPs 抑制剂对 XIAP-BIR2 有较高的选择性,因此有可能利用对 XIAP-BIR2 有较高选择性的化合物设计选择性降解 XIAP 的 PROTACs,从而实现对XIAP 的选择性抑制。这类化合物在阐明不同的 IAPs家族蛋白的生物学功能方面将会有广泛的应用。
综上所述,BIR2 选择性 IAPs 抑制剂是一类新型的 IAPs 抑制剂,不仅可以用于以 IAPs 为靶点的新药研发,而且可以用于 IAPs 的生物学作用机制的研究中。
来源:药学进展
原文链接:http://mp.weixin.qq.com/s?__biz=MzA5MDY3ODExNQ==&mid=2651320475&idx=1&sn=6b0cc93cccbdb9d2b028966c331b8438
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
三价镓化合物

化合物半导体的机遇

科学家创建新化合物库,有望预防常见手术
高温金属间化合物
插入化合物

【“长江学者”20年】药学院张勇慧教授:自然界中寻找创新药物
合熔化合物
化合物半导体材料
微晶金属间化合物
中国海洋大学在海洋天然产物生物合成机制研究方面取得重要进展


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号