科技工作者之家
科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
科技工作者之家 2020-11-29
来源:BioArt
近日,Genome Research在线发表了中国科学院上海生科院营养健康研究所计算生物学重点实验室邵振课题组的方法学论文MAnorm2 for quantitatively comparing groups of ChIP-seq samples,报道了其开发的新一代MAnorm2计算模型。该模型能够对多样本ChIP/ATAC-seq数据按照特定标签分组进行统计建模和组间定量比较,可靠地在样本组层面鉴定组间显著差异的ChIP/ATAC-seq信号。

染色质免疫共沉淀测序(ChIP-seq)实验被广泛用于刻画转录因子结合和组蛋白修饰的全基因组分布。比较来自不同细胞类型的ChIP-seq样本是刻画细胞分化和病变过程中动态转录和表观调控的关键基础。2012年,邵振与张一婧等合作在Genome Biology发表了用于两个ChIP-seq样本之间进行一对一定量比较的MAnorm模型。近年来,随着实验技术的发展和测序成本的不断降低,在ChIP-seq样本组(而非单个样本)之间进行比较分析已成为越来越常见的研究需求。一方面,研究者会产生同一实验的多个生物学重复来提高实验结果的可信度。另一方面,通过将来自不同个体的样本根据特定标签(如年龄、性别、患病与否、疾病亚型等)分组进行比较,研究者能够控制个体差异造成的影响,更可靠地识别与该标签关联的差异结合位点。然而,由于ChIP-seq实验固有的高复杂度和高噪声水平,以及不同比较场景所特有的技术困难,现阶段对多样本ChIP-seq数据进行分组定量比较仍然是一个巨大的计算方法学挑战。
在ChIP-seq数据标准化这一步,MAnorm2沿用了MAnorm的核心假设(即当两个样本间存在大量共有峰(common peaks)时,认为它们在这些区域的信号强度总体上没有变化,进而通过绘制传统M-A图并对共有峰M值和A值之间的相关性进行线性拟合来获得全局峰信号强度标准化的参照模型),通过重构其信号强度变换体系,新发展了以参照样本为基准的多样本并行ChIP-seq信号标准化流程。进一步,针对多样本分组比较的需求,MAnorm2搭建了一个理论上适应任意树状分组结构的层级化多样本标准化策略:首先分别对每个样本组适当地选取参照样本进行内部标准化,再向上一层把不同的组标准化至互相可比,以此来减少标准化过程中产生的偏差(biases)并提高结果的稳定性(图1)。
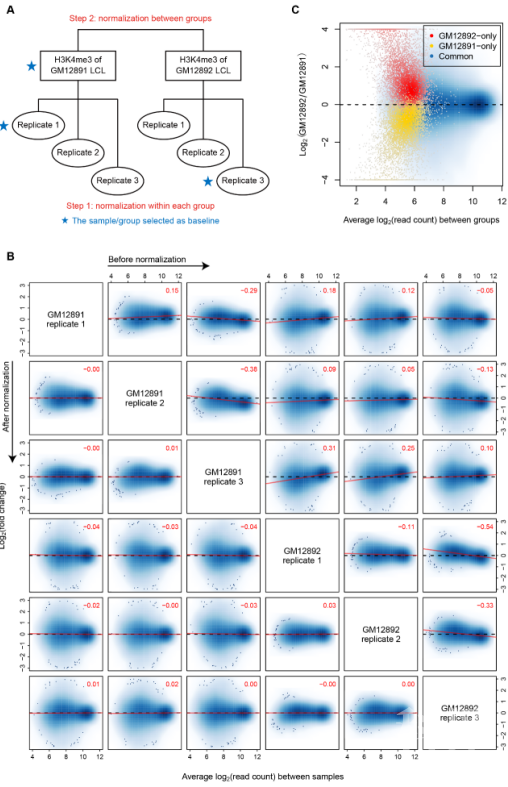
图1:(A)ChIP-seq样本组分层标准化的示意图。(B)每一对ChIP-seq样本在它们的共同峰区域上(标准化前和标准化后)的MA图。(C)两组ChIP-seq样本之间的MA图。
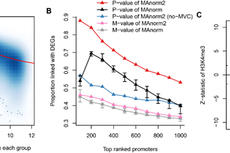
在完成标准化后,MAnorm2接下来针对每个基因组区域上观察到的ChIP-seq信号组间差异进行统计检验。在通常组内样本数较少的局限下(2-3个重复本),为了能更准确地衡量每一个基因组区域上的组内样本间ChIP-seq信号变化水平(within-group variability),MAnorm2通过拟合一条光滑的均值-方差曲线来实现不同区域间的信息互享(图2A)。进一步,MAnorm2设计了一个经验贝叶斯(empirical Bayes)框架,利用拟合好的均值-方差曲线来给单个区域的组内变化水平赋予一个先验分布。基于这个框架,MAnorm2可以通过平衡先验和后验观测来更准确地估计ChIP-seq信号的组内变化水平,从而提高对组间差异ChIP-seq信号的灵敏度(图2B、C)。
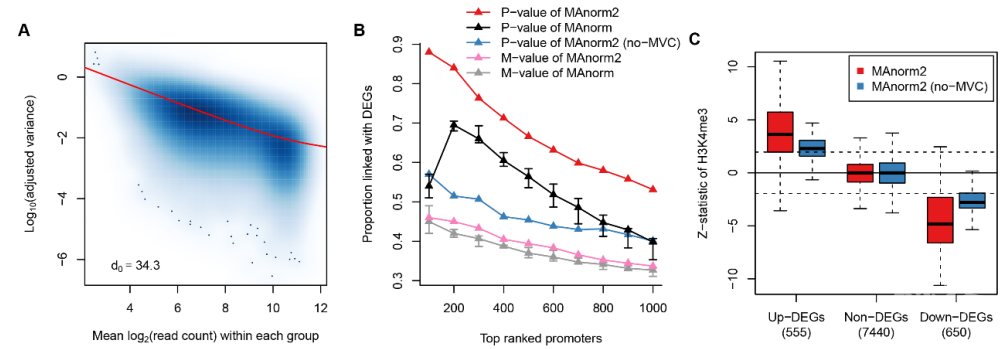
图2:(A)在不同基因组区域间拟合均值-方差曲线(mean-variance curve;MVC)。(B)根据不同的统计指标对基因启动子按照差异H3K4me3 ChIP-seq信号的可能性进行排序,并计算其中差异表达基因(differentially expressed genes;DEGs)启动子所占的比例。(C)检查不同类型的基因启动子上差异H3K4me3的统计显著性。虚线对应P值为0.05。
与已有的其他经验贝叶斯方法相比,MAnorm2最大的优势在于考虑了不同样本组的组内ChIP-seq信号变化水平可能存在系统性差别。这一情形在正常人和癌症患者之间的比较中经常出现:由于肿瘤组织或血液样本本身的异质性以及癌症亚类型和不同患病阶段的多样性,癌症样本组的组内信号变化水平往往显著高于正常样本组(图3A,B)。为了解决这个问题,MAnorm2通过在建模过程中引入一个方差比率因子(variance ratio factor),首先把不同样本组的全局组内信号变化水平修正至一致,然后使用修正后的方差进行均值-方差曲线的拟合(图3C)。
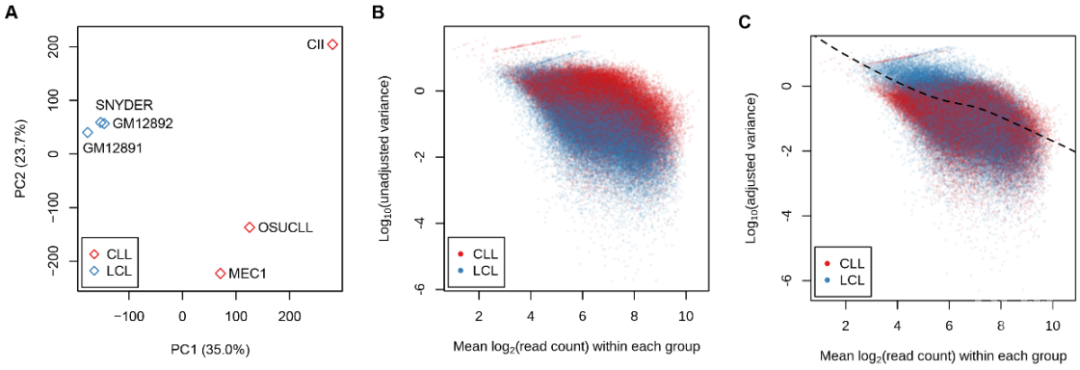
图3:(A)对来自不同的人的H3K27ac ChIP-seq样本进行主成分分析。这里LCL(lymphoblastoid cell line)组包含三个源于正常人的B细胞的细胞系;CLL(chronic lymphocytic leukemia)组包含三个源于慢性淋巴细胞白血病患者的B细胞的细胞系。(B)关于来自不同组的均值和未修正的方差的散点图。(C)关于均值和修正后的方差的散点图,以及由此拟合出的均值-方差曲线。
研究人员将MAnorm2与现有的其他ChIP-seq差异分析工具进行了系统的比较,发现MAnorm2展现了明显更优越的使用性能,尤其是当进行比较的样本组拥有明显不同的组内变化水平时,例如癌症和正常样本相比较(图4)。
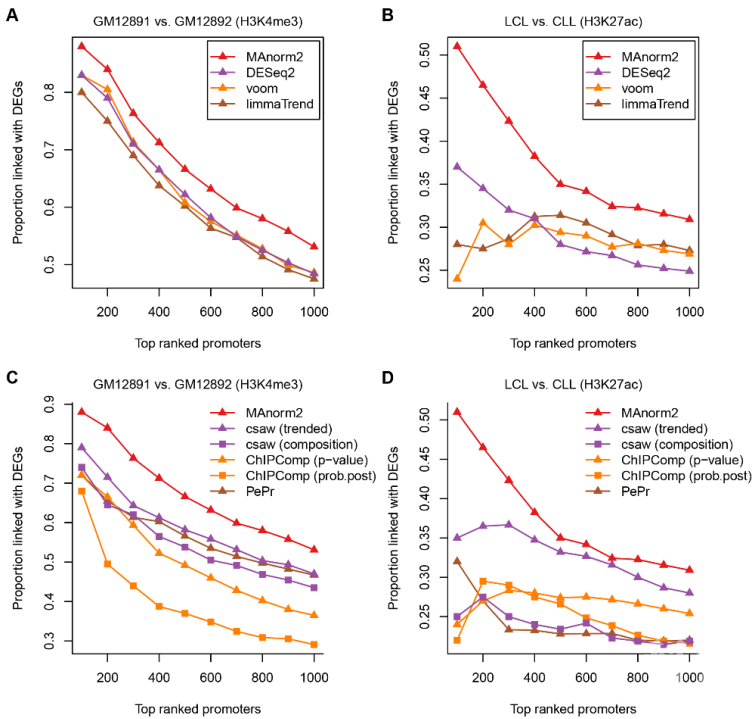
图4:(A,B)将MAnorm2与其他经验贝叶斯方法进行比较。(C,D)将MAnorm2与其他ChIP-seq差异分析工具进行比较。
MAnorm2模型的应用场景和统计模型具有良好的可扩展性。在正文中,研究人员不仅展示了MAnorm2在ATAC-seq数据差异分析上同样适用,还将其统计模型扩展到可以同时比较任意多个样本组,并发现其使用效果优于传统的ANOVA方法。
中国科学院上海营养健康研究所博士后涂世奇为该论文第一作者,邵振研究员为通讯作者。
来源:BioGossip BioArt
原文链接:http://mp.weixin.qq.com/s?__biz=MzA3MzQyNjY1MQ==&mid=2652511812&idx=8&sn=019f751aa27adae75969e736514fed1f
版权声明:除非特别注明,本站所载内容来源于互联网、微信公众号等公开渠道,不代表本站观点,仅供参考、交流、公益传播之目的。转载的稿件版权归原作者或机构所有,如有侵权,请联系删除。
电话:(010)86409582
邮箱:kejie@scimall.org.cn
中国农业大学杨淑华/施怡婷团队通过ChIP-seq揭示CBF转录因子的全基因组结合位点
单向方差分析
营养与健康所等建立MAnorm2计算模型
沈阳生态所在玉米农田肥料氮利用效率和损失研究中取得进展

中国科学院水生生物研究所关于广布性冷水鱼类温度适应机制研究取得新进展
多元方差分析
方差因子

中国农业大学杨淑华/施怡婷团队通过ChIP-seq揭示CBF转录因子的全基因组结合位点

抗疫宅家 临汾市【线上流动科技馆】第十四期
何爱彬组开发高通量、普适性单细胞ChIP-seq技术——CoBATCH


科技工作者之家APP是专注科技人才,知识分享与人才交流的服务平台。
 微信
微信
 京公网安备11010202008424号
京公网安备11010202008424号